Способ получения производных 13-галоидмилбемицина или их солей, или их сложных эфиров
Номер патента: 1235
Опубликовано: 15.09.1994
Авторы: Норитоси Китано, Акира Нисида, Кацуо Сато, Тосиаки Янаи, Бруно Фрай, Энтони О" Салливан









Формула / Реферат
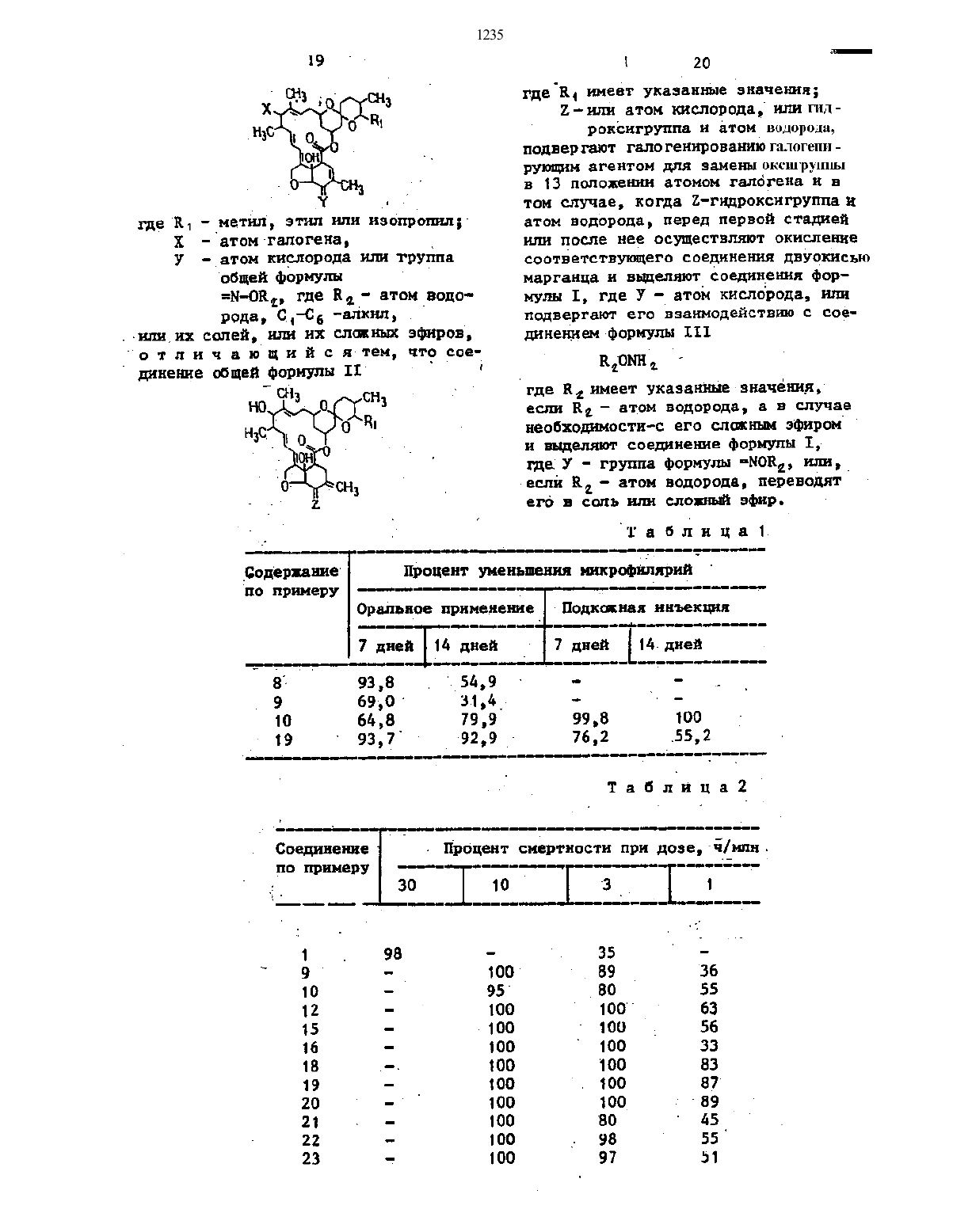
Изобретение относится к гетероциклическим соединениям, в частности к получению производных 13-галоидмилбемицина фор-лы I
лы =N-OR2, где R2-Н, С1-С6 - алкил, или их солей, или их сложных эфиров, которые проявляют антигельминтную, акарицидную и инсектицидную активность.Цель - разработка способа получения новых более активных соединений. Получение их ведут галогенированием соединения фор-лы I, где R1 указано выше, в положении 5- O или ОН и Н, а в положении 13-ОН галогенирующим агентом для замены оксигруппы в 13 положении атомом галогена и в том случае, когда в 5 положении ОН и Н, перед или после первой стадий осуществляют окисление соответствующего соединения двуокисью марганца и выделяют соединения фор-лы I, где У - О, или подвергают его взаимодействию с соединением фор-лы R2ONH2, где R2 указано выше, если R2-H, а в случае необходимости с его сложным эфиром и выделяют соединение I, где У - группа фор-лы = NOR2, или, если R2-Н, переводят его в соль или сложный эфир.
Текст
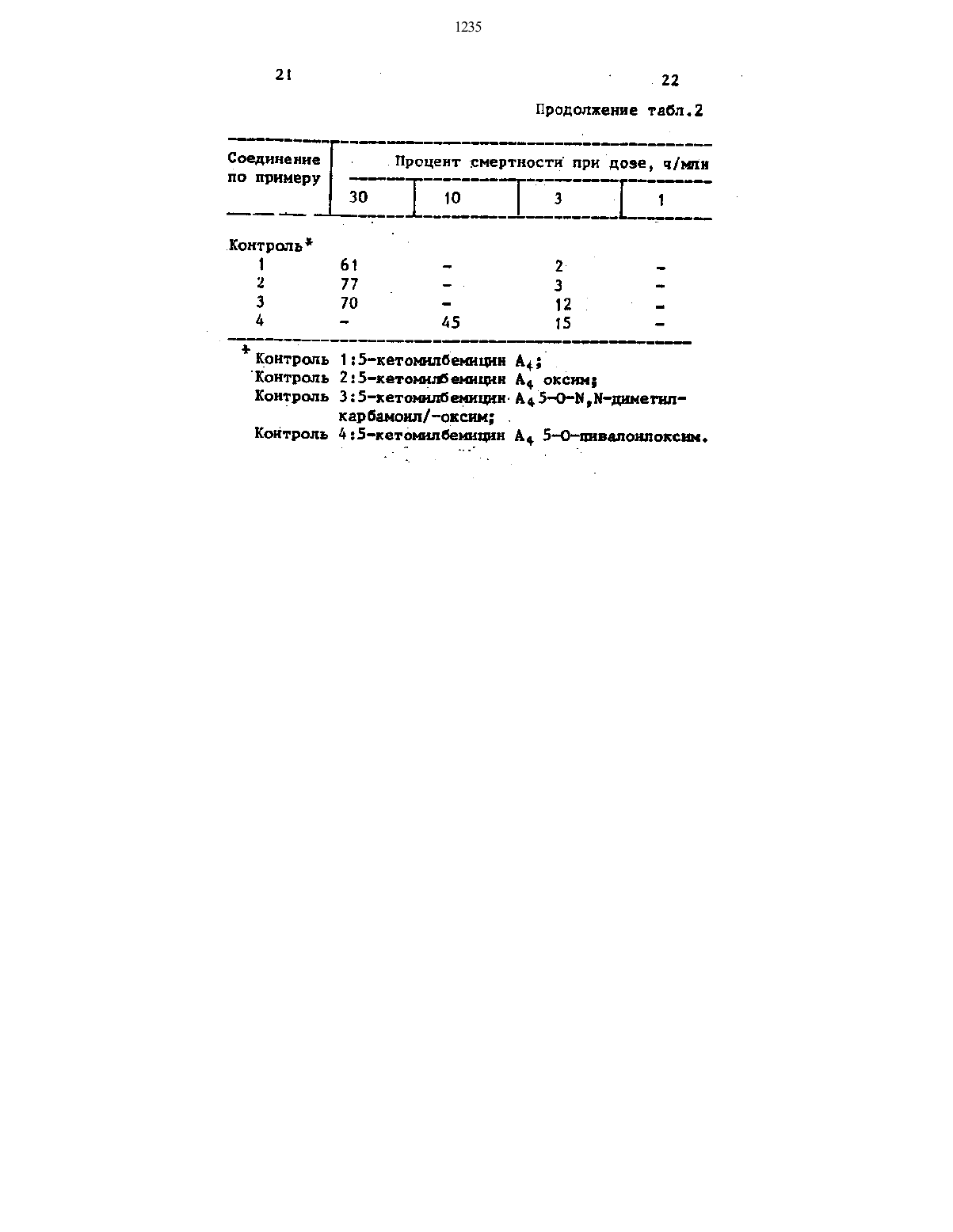
ддющх более высокой акарнцнднои активностью Способ иллюстрируется следующи ипрнмерамнМнлбеммцнны отвечают формуле Ан, нзопропил (шшбешашш д), Зтн соединення.могут быть выдепеч ны из культур Зсгерпошусее Штампа в-41-146.70 мг диэтиламмносульфотрнфторнда добавляют по каплям к раствору 560 мг 13 оксн 5 кетомилбенцина Ад в 25 мл метнленхлормда поддерживая темератУРУ при 60 С, н смесь перемеи вают в течене 15 мин. В конце этого времени реакцонную смесь вливают в воду н экстрагируют этилацетатом. Органический экстракт сушат надбез водным супьфатон натрия н концентрируют выпариванем при понненном47 мкл тноннллорнда добавляют по каплямк раствору 235 мг 1 З-оксн 5 кетонпбемцннаА,в 40 мл сухого бензола в условиях охлаждения льдоми смесь перемешивают при комнатной температуре в течение 2 ч. Реакционную смесь затем вьшивают в воду и затем обрабатывают таким же способом, как описано в приере 1, с получением 100 мг (выход 612) целе вого соединения. Массспектр (ш/т) 574 (М), 556,538. Шт-спеэятр (свс 1,Б ч/мнн 4,12Повторяют методику, описанную примере 2, за исключеннем того, что были нспопьзованы 280 мг 13 окси-5 кетомнлбемнцмна А,.и 80 мг трибромшщ фосфора, С получением 90 мг (выход 292) целевого соединения.. Инфракрасные спектры поглощенияд освг) 1,см- 3450, 1715, 1 вво.45 мг трнетмлснннллорнда добавляют в атмосфере потока азота к раст вор 194 нг 13 окси 5 кетомлбеми цнна А, н 66 мг нодида натрия в 151 ш ацетонитрила, смесь перемешвают при канатной температуре в течение В ч. Реакционную смесь выпивают в воду и затем обрабатывают таки же способом, как описано в примере 1, с получением 96 мг (выод 412)целе вого соединения.инф акрасные спектры поглощения ног диск эдьо, 1735, 1715,1685.П р и м е р 5. 13 р-Фтор-5 кето нпбемцин В. 0,60 г активированной двуокиси марганца-добавляют к раствору 170 мг 13 фтормилбеицнна В 3 3 мл метн ленхлориде при комнатной температут ре, н смесь энергично перемешивают в течение 10 мин н нерастворимую честь промывают метмленхпормдом. Фильтрат и промъочный растворы объ 5единлют н концентрируют выпариванием при пониженном давлении. Остаток очищают на колонке с силикагелем с получением 151 мг (выйод 891) целевого соединения.(полученного как описано в примере 5) н 36 мг гидрокснламна гидролорида добавляют к 3 мл этанола, и смесь перемешивают при 0 С в течение 90 мин. Затем реакционную смесь охлаждают и концентрируют вперилаиием при пониенном давлении. К остатку добавляют бензол и воду удаля ют авеотролно 1 п айвы. Остаток очи щают на хроматографической колонке с силикагелем с получением 90 мгП р и м е р 7. 13 милбемицн В окснм. 119 мг 1 Зхлорнилбемнцина Вн 0,80 г активированной двуокиси мар ганца реагируют таким жеобраэом как описано в примере 5, с получением 13 р-хлор-З-кетоюшбешцзана В,46 мг гидроксиламмн гидрохлорида добавляют к реакционной смеси и всю смесь обрабатывают таким же образом,как описано в лримере 6 с получением 97 мг (выход 802) целевого сое дмнения. Массспектр (ш/г) 603 (М).П р и м е р 8. 13 р -хлор 5 кег томлбемции Ад оксим.39 мг 13 клормнлбемицнна Ад,0,10 г активированной двуокиси марганца и 10 мг гидроксиллии гидро хлорида реагируют таким же образом,-как описано в лринере 7, с получением 15 мг (выход 382) целевого соедмненил.Раствор 268 мг 13 чфтор 5 кетомилбемицнна Ае (полученного как описано в примере 1) в 4 мл метанола и 4 мл диоксана добавляют по каплям к. раствору 166 мг гидроксиламн гидрохлорида в 3 мл воды, и смесь перемешивают при комнатной температ ре в течение 8 ч. После этого времени реакционную смесь вливают в воду и экстрагировали этилацетатом. Экстракт сушат над безводным сульфатом натрнл.и затем концентрируют выпариванием при пониженном давлении. Остаток очищают на хроматографической колонке с снликагелем с получением 182 мг (вывод 66,42) целевого соедннения.103,8 мг 13 хлор 5 кетомипбемици на А (полученного, как описано в примере 2) и 75 мг гидрокснламин гидрохлорида обрабатывают таким же способом, как описано в примере 9, с получением 63,4 мг (выкод 59,52) целевого соединения.68 мг 13 иод-5-кетомлбемицна А, н 35 мг гидрокснламин гидрохлорида обрабатывают таким.же способом,как описано в примере 9,сполучением 48 мг (выход 692) целевого соедииения.рИод-5-кето 1235 7. 129 мг смеси 2,31 по весу 13 фтор-Ззкетоммлбемицина Ад и А, и 115 мг 0 метилгидроксиламин гидро- р хлорида реагирует таким же способом,как описано в примере 9, с получат ннем 108 мг (выод 80)целевого соединения. . Инфракрасны спектр поглощения дквг) 4 см- з 47 о 1715. ЯМ-спектр СВС 1 д) 8 ч/млн 3,917,7 мг 1,4-диазобицикпо (22,2)е октав и 5,4-мкпацетнлпорида добавляют к раствору 40,5 мг 13-ипор 5 кетомпбемцина Ад оксид Спел ченного как описано в примере 10),в 1,5 мл ацетонитрнпа, и смесь перемешивают при комнатной температуре в течение 6 ч. После этого реак пшенную смесь вливают в воду н экстрагируют этилацетатом. Экстракт суфат над беэводнм сулъфатом натрия и концентрируют выариваннем при пониженном давлении. Остаток очищают на хроматографической колонке сА, оксима полученного как описаноП р н м е р 17. 1313 Хпор-5 ке тонлбемицин Ад 5-О(Ыметилкарбамонп)оксин.0,30 мл метнпнвоцанатв добавили к раствору 100 мг 13 клор 5 кетомлбемцнн А, оксиа (полученного как описано в примере 101 в 2 мл тетра.гндрЬФурене, и смесь оставпнют стоять в течение 8 ч, оставляя ее герметично закрытой.После этого растворитель дистиллруют при пониженном давленн с получением 95,7 мг (выход 871 целевого соединения.57 мг 13-фторч 5 кетоипбемцина А 4 окснма (полученного как описано в примере 9) и 12 мкл Ы,Ыднметип карбамоип хлорида обрабатывают таки же способом, как описано в примере 15, с попученнемд 9 мг (вывод 761) целевого соедмнения.15 мл триэтнламна и 14 мкл пи- П р и м е р 23. 13 р-Фтор 5 ке аалонлхлорида добавляют к раствору томилбемицин А, 5 Опропонлокснн,57-мг 13 рфтор 5 кетомшпоемцина А, Повторякп методику примера 15,оксима (полученного как описано Е но с исполъэоианием пропионнллорн примере 9) в 10 мл бензола н смесь да, с получением целевого соединения. затем перемешивая при комнатной Масс-спектр (Ш/г) 645 (М)589. температуре в течение 3 ч. После ЯМР-спектр (свс 1,)8 ч/млн 41,11 этогореакцнониУю смесь обрабатыва- (1 н дуплет, 3 10,4 Гц) 4,61. ют таки же СПОСОЙОН. Как ОПНСЗНО (1 Н, синглет) 4,96 (ТН, сннглет).. Массспектр (Щ/г) 657.(М) иилоксим. 639,574. . Повторяют методику примера 15, ЯМтспектр (СВС 13)8 Ч/млн 1,29 но с испольэованем 1315 фтор 5 ке(стадия Е). яМтспектр (СВС 1 д) ч/млн 2,4 д Повторяют методику ПРИМЕРЕ 15. ц д(3 Н синглет) 3,84 (1 Н синглет) но с использованием октаноил клори д,и 1 (1 н дуплет дуадетд 3 5 1 оода и снеди 13 Э-Фтог-Б-кетошлбеш- и 47,2 Гц) д,54 (1 н, сннглед). ЦННОВ А 4,5 оксимов (полученным по П р и м е р 25. 13 р-Фтор 5 кеМетодике ПРННЕРЯ 9), с получением томнпбемнцин Ад 50(пентаацетнп смесн 2,011 целеаогосоедннения. глюжоноил)оксим. Нассспектр ш/г) 699 (н),685. повторят методику, описанную вЯФ-спектр (СВС 13) 3 ч/нпн 4,41 примере 15, но с испольаованем (1 Н, дублет дублетов Д 10,1 и 13- фтор 5 нетоммлбемицин А, оксид 3 д ГЦ) 4,53 (1 Н синглет) 4,8 на (полученного как описано в при(1 Н, широкий синглет). - - мере 9) н пентаацетнлглюконил хлоП ри м е р 21. 13 Ь-Фтор 5 кето рида с полученем целевого соедннпоеммцины А 55 О-гексадеканонлок- ценил. . сим. Массспентр (ш/г) 615 (М 3 д 6),Повторяют методику примера 15, но 573. с использованием гексадеканоип хлоч яМтспектр (СВС 13)Б ч/млн 3,90 рида и смеси 13 фтор 5-кетомнпбемн (1 Н, синглет) д,1-4,д 5 Н, мульдни А 5 окснов (полученных по ме типлет) 4,52 (1 Н, синглет). топике пример 9), с полученем сме П р н м е р 26. 13 Бром 5 кето си 1,О 1 целевых соединений. ммлбемцин Ад. - Массспектр Сш, А 4) 811 (Н). 20 мкл брометилсилана по каплям янтспектр (СВС 1,)8 чмн 3,95 добавляют при охладенн льдом в(18, синглет) 4,43 (1 Н, дублет ду раствор 27 мг 13 гидрокси 5 нето Бпетов, Д 9,9 и 48,0 Гц), 4,58 мнлбемицина А, в 15 мл сухого мети(1 Н синтлет). . пенлорнда, а затем смесь переменП р н м е р 22. Кр-Фтор 5 кето- воют при комнатной температУрев тенлбенцин А 4,3 5-О-ацетилоксны. чеиие ночи. Реакционную смесь вьшщ Повторяют методику примера 15, ватт в воду и подвергают обработке но с использованием смеси 131 Эфтор по методикепримера 1, получая 52 мг 5 кетомнлбемцин А 4,д оксимов (полу- (19,2 Х) целевого соединеия. ченнык по методике пример 9), с полу- П р и м е р 27. 38 мкл метилйодида ченем смеси 1,81 целевых соедине- по каплям добавляют в 104 мкл трит кий. п . фенлфосфнта при комнатной темперанасс-спектр (Щ/г, Ад) 615 (М). туре и смесь перемешиваютв течение ЯМ-спектр (СВС 13) ч/млн 3,96 1 ч. Ксмесн,-разбавленной 2 Н Не г
МПК / Метки
МПК: C07D 493/22, A01N 13/02
Метки: солей, эфиров, способ, сложных, получения, 13-галоидмилбемицина, производных
Код ссылки
<a href="https://kz.patents.su/11-1235-sposob-polucheniya-proizvodnyh-13-galoidmilbemicina-ili-ih-solejj-ili-ih-slozhnyh-efirov.html" rel="bookmark" title="База патентов Казахстана">Способ получения производных 13-галоидмилбемицина или их солей, или их сложных эфиров</a>
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты или их фармацевтически приемлемых солей
Номер патента: 1232
Опубликовано: 15.09.1994
Авторы: Эйити Косинака, Нобуо Огава, Нориюки Яги, Ясуо Итох, Хидео Като, Томио Сузуки
МПК: C07D 401/02, A61K 31/495
Метки: пиперазинилхинолин-3-карбоновой, способ, солей, фармацевтически, получения, приемлемых, 6-фтор-1,4-дигидро-4-оксо-7-замещенной, производных, кислоты
Формула / Реферат:
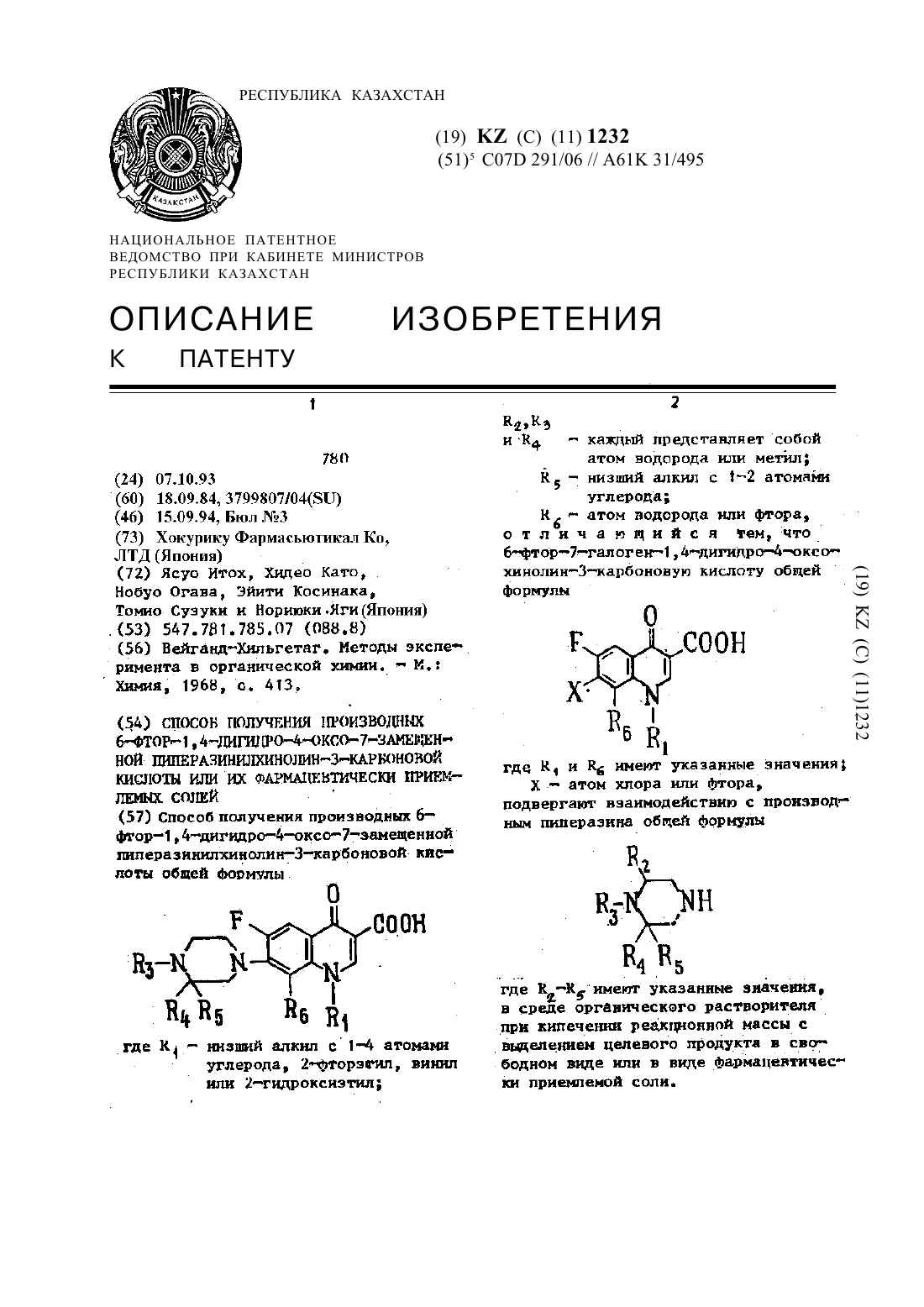
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты общей формулыгде R1 - низший алкил с 1-4 атомами углерода, 2-фторэnил, винил или 2-гидроксиэтил; R2, R3 и R4 - каждый представляет собой атом водорода или метил; R5 - низший алкил с 1-2 атомами углерода; R6 - атом водорода или фтора, отличающийся тем, что 6-фтор-7-галоген-1,4-дигидро-4-оксо-хинолин-3-карбоновую кислоту общей формулыгде R1...
Способ получения цис, эндо – 2 – азабицикло – ( 3, 3, 0 ) – октан – 3 – карбоновых кислот или их кислотно – аддитивных солей .

Номер патента: 1234
Опубликовано: 15.09.1994
Авторы: Рольф Гайгер, Фолькер Теетц, Хансйорг Урбах, Райнхард Беккер, Бернвард Шелькенс
МПК: C07D 451/02
Метки: азабицикло, кислот, цис, карбоновых, аддитивных, эндо, кислотно, способ, получения, октан, солей
Формула / Реферат:
Изобретение касается замещенных цис,эндо-2-азабицикло-(3,3,0)-октан-3-карбоновых кислот, в частности соединений общей формулыгде R1 - низший алкил, - (CH2)4NH2, СН2-С6Н4-n-0 - низший алкил, CH2C6H4-n-OH; R2 - радикал-алкил, бензил; Y - Н или ОН; Z - Н или Y+Z-кислород; Х - фенил, или их кислотно-аддитивных солей, которые обладают способностью снижать кровяное давление. Цель изобретения - создание новых замещенных кислот с фармакологическими...
Способ получения производных 3-окси-2-циклогексен-1-она

Номер патента: 1217
Опубликовано: 15.09.1994
Авторы: Гернот Рейссенвебер, Винфрид Рихарц
МПК: C07C 49/713
Метки: получения, производных, способ, 3-окси-2-циклогексен-1-она
Формула / Реферат:
Изобретение относится к циклическим кетонам, в частности к получению производных 3-окси-2-циклогексен-1-она формулы CH2-CHR1-CH2-C(O)-C = [С(О)R2] - С(ОН), где R1 - низший алкенил. С6 - циклоалкил, который может содержать одну олефиноненасыщенную связь, низший алкилтионизший-алкил, С7-12 -бициклоалкил, который может содержать две олефиноненасыщенные связи, фенил, незамещенный или замещенный низшим алкилом или галоидом, или низшей алкилтиогруппой...
Способ получения производных пиридина

Номер патента: 1233
Опубликовано: 15.09.1994
Авторы: Акира Нохара, Еситака Маки
МПК: C07D 401/12
Метки: пиридина, получения, способ, производных
Формула / Реферат:
Изобретение касается получения производных пиридина, в частности соединении общей ф-лы где R1 - водород, метокси или трифторметил, R2 и R3 независимо - Н или СН3, R3 - фторированный С2-С5-алкил; n = 0 или 1, которые могут быть использованы как противоязвенные средства. Цель - создание новых более активных веществ указанного класса. Их синтез ведут реакцией соединений ф-л где R1-R4 - см. выше; один из Х1 и Х2 - SH, другой - галоид. При...
Способ получения производных фенилпиридазина

Номер патента: 1224
Опубликовано: 15.09.1994
Авторы: Энгельберт Клоймштайн, Франц Ранингер
МПК: C07D 237/12, A01N 43/58
Метки: получения, фенилпиридазина, производных, способ
Формула / Реферат:
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФЕНИЛЛИРИДАЗИНА общей формулы:где R - линейный или разветвленный алкильный радикал с 1-18 атомами углерода, взаимодействием соли 3-фенил-4-окси-6-хлорпиридазина с алкилтиохлорформи атами формулы:где R имеет указанное значение, в среде растворителя, отличающийся тем, что с целью упрощения процесса, в качестве растворителя используют водно-ацетоновый раствор, процесс проводят при 5-60 °С и выделяют целевой продукт...
Предыдущий патент: Способ получения цис, эндо – 2 – азабицикло – ( 3, 3, 0 ) – октан – 3 – карбоновых кислот или их кислотно – аддитивных солей .
Следующий патент: Способ получения производных эрголина или их солей
Случайный патент: Устройство для остеосинтеза переломов шейки бедра