Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты или их фармацевтически приемлемых солей
Номер патента: 1232
Опубликовано: 15.09.1994
Авторы: Эйити Косинака, Ясуо Итох, Томио Сузуки, Нориюки Яги, Нобуо Огава, Хидео Като




Формула / Реферат
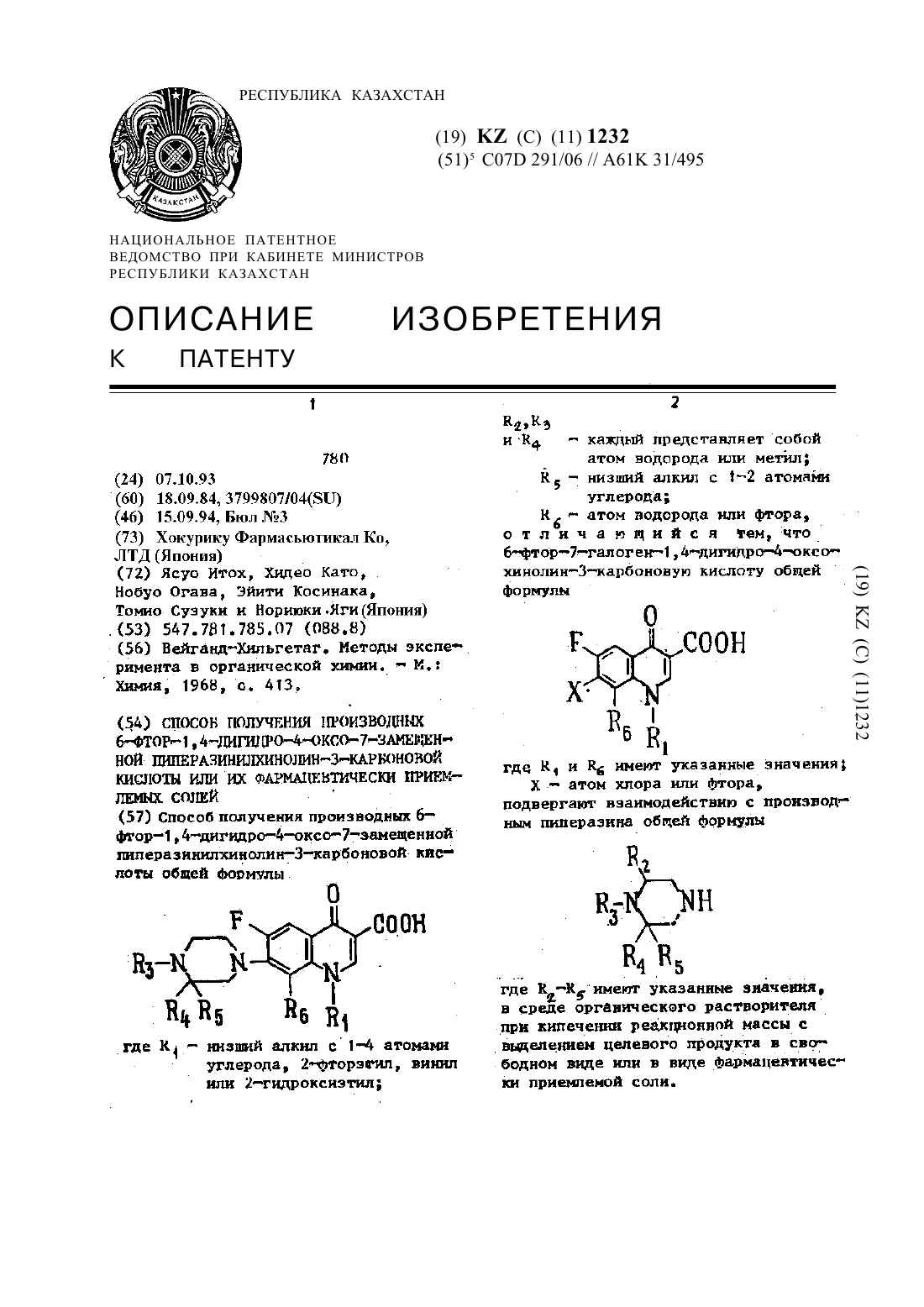
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты общей формулы
где R1 - низший алкил с 1-4 атомами углерода, 2-фторэnил, винил или 2-гидроксиэтил; R2, R3 и R4 - каждый представляет собой атом водорода или метил; R5 - низший алкил с 1-2 атомами углерода; R6 - атом водорода или фтора, отличающийся тем, что 6-фтор-7-галоген-1,4-дигидро-4-оксо-хинолин-3-карбоновую кислоту общей формулы
где R1 и R6 имеют указанные значения; X - атом хлора или фтора, подвергают взаимодействию с производным пиперазина общей формулы
где R2-R5 имеют указанные значения, в среде органического растворителя при кипечении реакционной массы с выделением целевого продукта в свободном виде или в виде фармацевтачески приемлемой соли.
Текст
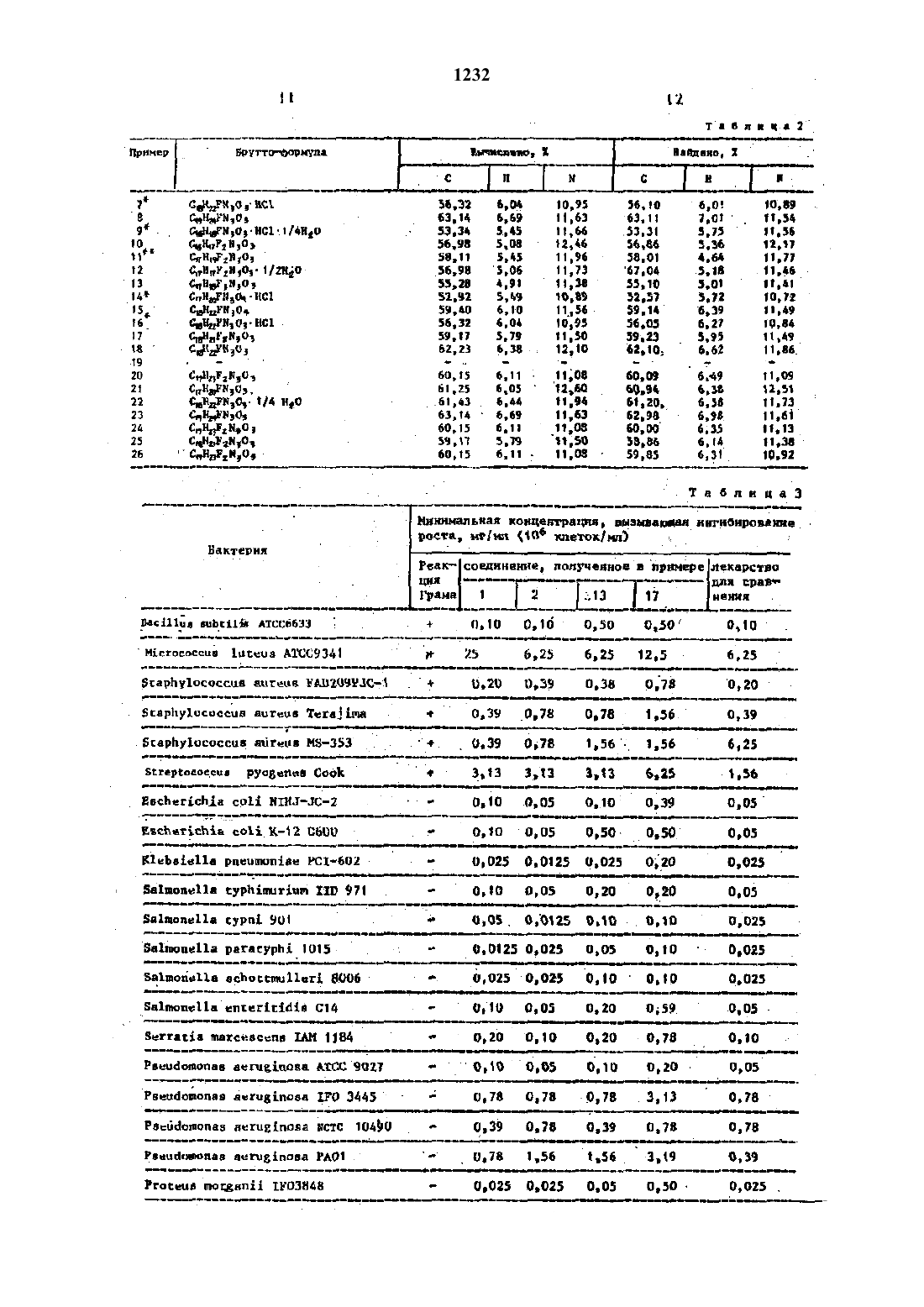
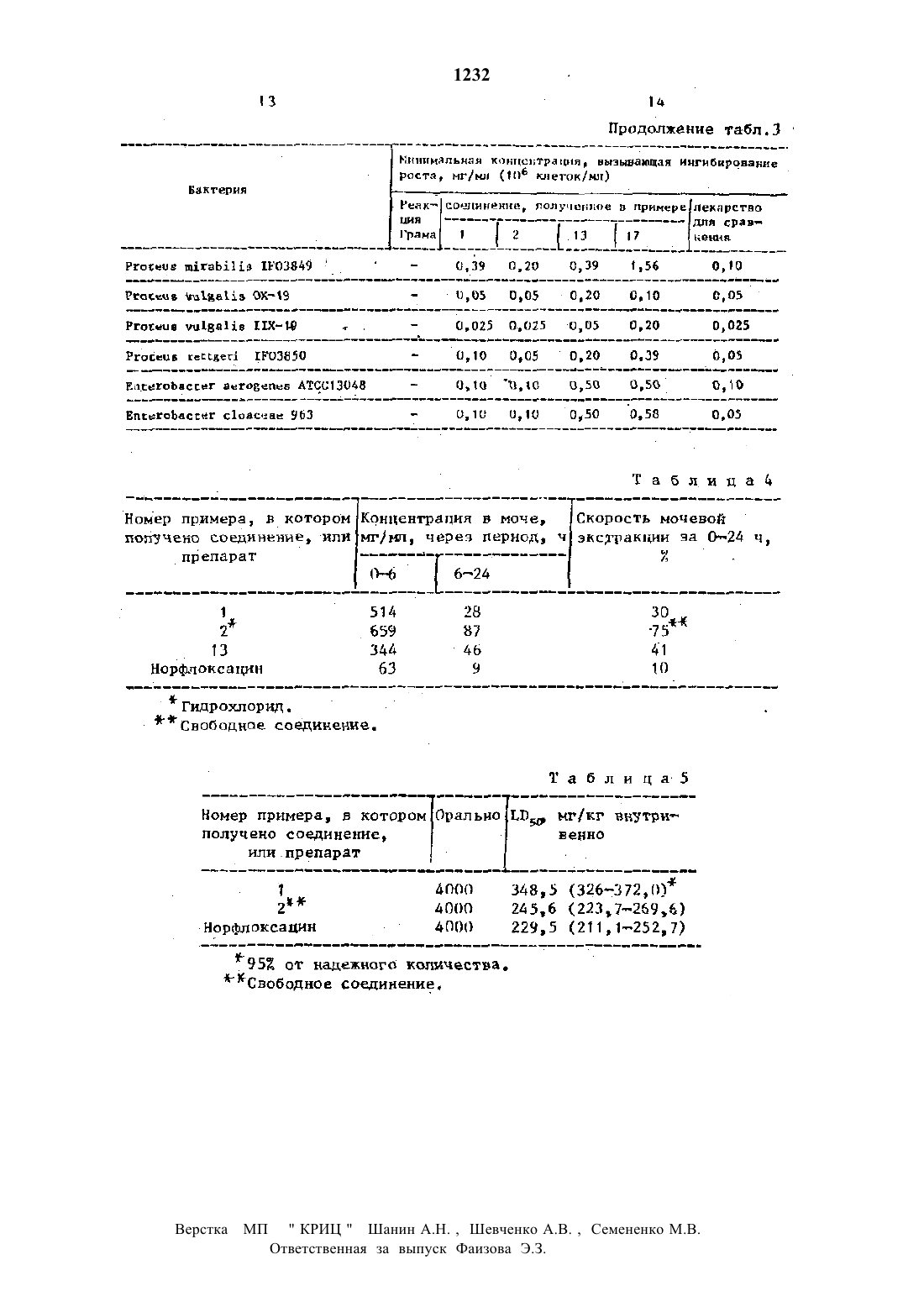
Изобретение относится к способу получения новыж производных бчфторт 1 цднгидроидпоксо 7 нзамещеннойлит перазиннлхинолинт 3 каР 50 Н 0 В 0 йКНСЛ 0 ты, обладающмх.сильнъш антибактерип альнм действием. . УЦель изобретении синтез новых производным пиперазинилхинопинтзт карбоновых кислот, по своей активи кости превосходящих известный препарат норфлоксацНн-нвлпющйся их стрэктурт ны аналогом. .Смесь 15,00 г 7 тнлор 1 тэтилт 6 т фторт 1,дтдигидротдтоксохинолинт 3 карбоновой кислоты, 16,70 г Зтметнля пиперазнна и 70 мл пиридина нагревают в течение 1 д чс дефлегммрованиен Растворитель реакционной снесивыш ривают, а остаток окисляют 502 нм водным раствором уксусной кислоты. Этот раствор-обрабатывают активированным углем-и нейтрализуют 20 тнын водным раствором гидроокиси натрия Затем раствор обрабатывают вновь акт тнвированным угленн концентрируют. Осадок отфильтровывают н растворяют в этаноле. Раствор окисляют хлористым водородом в этаноле и концентрируют Осадок отфнльтровывают и перекрнсталт лизовывают из водного раствора эта нола с-получением 8,19 г гидрохлорида целевого соединения в виде бледнот жепты игл, т.пл. 300 с.Смесь 1,00 г 1-этнлт 6,78 ттрифторн 1 дтднгидродиоксохннолин 3 ткарбонон вой кислоты, 1,10 г 2 тметилпгптеразина и 10 мл пиридина нагревают в течение 15 мин при дефлегнированни. Реакцнонт ную смесь выпаривают и к остатку дот бавляют метанол. Осадок отфильтровывают и перекристаллизовывают из этанолас получением 0,36 г целевого соединения в виде бесцветным игл,тпп. 239 н 2 д 0,5 с. 1Найдено, 1 С 57,98.Н 5,47 н 12,18. Обьмнын способом готовят гидрохлое рнд н перекрнсталлнэовывашт его из воды в виде бесцветныш кристаллов,т.пд 29 он 3 опс (раэл). Вычислено, 2 С 52,65 Н 5,20 Ы 10,84. СНдБ 2 Ы 303-НС 1 Найдено, 2 С 52,78 Н 5,32 н 10,65 ь- П р и м е р 3. 1 тЭтип 6,8 тдифторн 1,4 дигидрот 7 т(3 метилт 1 тпиперазинил) доксохинолин-Зткарбоновая кислота. К раствору 0,55 г этилт 1 этил 6,8 н днфтор 1,4 тдигидрот 7(3-метил 1 пипе разннил)доксохннолнн 3 чкарбоксилата н 5,5 мл этанола добавляют 11 мл 182 ной соляной кислоты и смесь нагревают в течение 4 ч при дефлегннровании. Осадок отфнльтровывают н промывают этанолом и эфиром. Перекристаллиэан цией из воды получают 0,43 г гидроп хлорида предлагаемого соединения в-виде бесцветныщ.игл. По реврльтатам ЯМР н Нктспектроскопии это соедине ние идентично соединению, полученному в примере 2. Ч Исходный этилт 1 нэтилн 6,8 дифтор 1,4 пдигидро 7 т(3 тметнлгТтпиперавнт пил)4 оксохинолннтзчкарбоксилат готовят следующим образом. Снесь 1,50 г этил 1 зтнлт 6,7,8 т трифторн 1 дтдигидротдтоксохинолинтвина и 5 мл пиридина нагревают в течение 3 ч при дефглегнмровани. Растворитель из реакционной снеси выпаривают н остаток растворяют в хлороформе. Раствор пронзают водой,сушат н выпаривают. Остаток перекрнсг талпизовывают из смеси бензола и изопропилового эфира с получением 1,00 г бесцветым игл, т.лп 126,5127,5 с-днгндро 1 нвопропилт 7 н(3-нетил 1 т пилеразинил)тоноксохинолии 3 карбоно вая.кнслота. Смесь 0,68 г 6,7,Внтрифторт 1,4 днгндро 1 тиэопропнл-4 оксохииолин 3 карбоновой кислотыП,72 г 2 метилпиг перазнна н-10 мл пиридина обрабаты с 60,30 н 5,345 вают, как в примере 2,-с получением 0,42 г бесцветный кристаллов целевоч го соединения, т.пл 217218 С Вычислено, 21 С 57,75 Н 5,92,Н 11,32. Найдено, 2 С 57,50 Н 5,97 11,13. 6,78 трифторг 1,ддигндрот 1-ивот пропилт 4 оксохинолинчзнкарбоновуюлна, 10,30 г ацетата натрия, 20 м ацетона, 19,6 мл уксусной кислоты и 39 мл воды при перемешивании и охлаждении льдом. Спустя 2 ч смесь подщелачнвают карбонатом натрия и вкстрагнргютбензолон. Экстракт промывают водой, насыщенной хлоридом натрия, сушат н после выпаривания растворители получают 3,17 г 2,34 т трифторттгиэопропилаиилнна в виде бесцветного масла.Смесь 2,50 г 2,З,амтрифторМтнзон пропипанилииа и 2,80 гдиэтил 2 г этоксиметиленмалоната нагревают в течение 1 ч при 16 О 170 С. К реакцин онной снеси добавляют гексан, а затем охлаждают. Отфльтровывают кристаллы с получением 2,45 г днэтн 2 н 2,З,4маловата, который перекристаллизовыт воют из гексане в виде бесцветныч игл, т.пл. 92,593,ПС.Смесь 9,00 г дизтилп 2 г(23,4 н трнфторПНнзопропипанилино)нетиленп малоната и 90 г попифосфорной кислоты нагревают в течение 1.ч при 8085 С с перенешиваннем.Реакцнониую смесь выпивают в ледяную воду и экстгвышагивают. Смесь 90 мл 182 тной со ляной кислоты и 45 мл этанола добавч ляшт к.остатку и дефлегнмруют в те чение 1,5 ч. Осадок осфилътровывают и прощают низ смеси хлороформа н этанола в виде светлочкоричневых игл, тпл. 261 т 262, 5 с. - - - Вычислено, 2 С 54,7 д Н 3,53 Н 4,91. цдтдгзно, . . Найдено, С 34,64 Н 3,7 Ы 4,53 П р и м е р 5. (К)1 г 9 тил 68 днфторт 1,ддигидрот 7(3 метнл 1 тСмесъ 20 П г 1 пэтил 6,78 трнфторн 1.дтдигидротдтоксохннолннЗткарбоно войцкислоты, 1,50 г (н)тг 2 метил пиперазинаы б,д (с 1, этот вол), и 15 мл пиридина нагревают в течение 15 мин при дефлегмнроваиии. после завершения рванин растворитель выпаривают и остаток растворяют В 10 тной соляной кислоте. Растворнейтрализуют водным раствором бикарчют, сушат и растворяют в смеси хлоп рофорна и метанола. Раствор окисляют соляной кислотой в этаноле. Осадок отфильтровывают и промывают в воде. Раствор нейтралнэукп-водньвараствотровывают с полрчениен 1,72 тпредлшп гаемого соединения, которое перекрнс таллнэовали из смеси хлороформа н этанола в виде бесцветных игл, т.пл. 2445-2 ь 5,5 со з 95 (с 1 хлороформ). Р вычислено, Х С 58,11 Н 5,д 3 н 11,96. .Найдено, 7 с 58,12 н 5,72 Н 12,07.-Смесь 1,50 г хлорс 6 тфторп 1 т(2 фторэтнлт)1,4,пднгидромдтоксохинолинг Зткарбоновой кислоты, 1,60 г 2 тнетил пиперазина и 8 мл пиридина нагревают в течение т 1 ч-при дефлегммрованни. Растворитель из реакционной снеси выпаривают и остаток растворяют в горячей воде. После охлаждения раствора отфильтровишают осадок и передкристаллизовывают его из этанола -сВ та 5 л.2 приведены данные злеиентп ного анализа синтезированным соедии нений. --нипераяина и 5 ил пиридина нагревают в течение 20 мин при дефпегмированни. Реакционную смесь выпаривают, а остаг ток растворяют в снеси хлороформаи иетанола. Раствор окисляют соляной кислотой в этаноле. Затем осадок отт фильтровыващт и растворяют в воде. Раствор нейтрализуютбикарбонатом натриии экстрагируют хлороформом. Экстракт промывают водой, сушат и выпаривают. К остатку добавляют метат ноп н остаток отфильтровывают сподучениен 730 мг требуемого компонента, который перекрнсталлизовншают на смеси хлороформа и этанола ввидедля соединений, полученным в прим мерах 1 и 2 (свободная кислота), 13 и-17, определяли антибактериальную активность. Для сравнения использова ли лекарственный препарат иорфлоксап ни.Минимальные защитные концентрации определяли способом двукратного рази равнения отара. Культуры на ночь сует Ьендировали в бульоне Ице 11 ех Нйпьоп с помощью соленого желатина. Одну 6 порцию бактериальной суспензии (10 или 108 образующх.колонюединиц/ми) инокулировали на пластинах, содержат-щх исследуемое соединена. Пластины инкубировали в течение 18 ч при 37 С. Минимальной защитной концентрапней была наименьшая концентрация соединения, ингибирующая заметны рост. Полученные результаты приведены в тавлдз.Для определена мочевой экстракт. ции самцов крыс-рода 0 весом 180 н 210 г разбили на группы по восемь авотнык в каащой. Иссцедуене соедин нення суспеидиовали в 0,52 тиом расти воре карбокснметилцеллюловы и вводили через рот крысам,которын в течене 24 ч перед этим не давали есть, в дозе 20 нтЧкг. соорщшиочэиаа пронес-ч жутки 06 и Бт 2 д ч и измерили кочевую екстракию путем биологического анат пиво с использованием Евеьетйспйа сон НЩНШ 1 С-2 Результаты приведены табзъда .Для биологического анализа применяли способ с использованием Еасьегйв сЬйа со 11 н 1 н 1 -1 С-2 . Перед началом мочу,в слтчае необходимости, равбавт лили 11.15 н фосфатиын буферным расти воромс рн 7,0для определения острой токсичности использовали самцов мышей рода аду и возрасте четырех недель по 10 жнннотных в группе. Исследуемые соединен нин суспенднровалн в ДЫР-нон раса-вот ре нарбоксинепшцехшюпозы и вводили юнцам аврально. В случае внутривенного введения исследуемые соединения раст-чворндш в 0,1 н растворе нс 1 и ней-ячрализовалн 0,1 н. растворен ИЗОП 1.1) определит по способу Рто Ьос от мертвых пластинах через 10 дней Полученные результат приведены в табл.5. .Как видно из прнведеншопгншче ре зультатов, соединения данного изобрет теннн проявляют сильный эффект монетрвой экстрактор и значительно меньшую дгокснчностъ по сравнению с эталонным лекарством. Кроме того, антибактериальныйданного изобретения примерно равны эталонному лекарству.Таким образом ясно, что воедино-о эшя данноголзобретенин являются весы-ш полезные-ш увекарстваш дляшинического использования благодаря их фарнаколотнческомъгдействию, а именно прекрасной абсорбции-н малой токсичности.Благодаря этому соединения предлатексного изобретают ванного безопаснее, .чем традиционно выпускаемое лекарство, н значительно более эфдфектнвнн в качестве клинического срсдства ы Необходимое - количество. для лечения обычно следует вводить оралъво всего 150-4000 мг отделвнъш дозами 2-4 разв в день на одного взрослого человека.каждыйнредстанпяет собой атом водо.Рода впитает-гл К т- низшй алкил с 1-2 атомами углерода н кв деток водорода или фтора.кристаллик Бесплатная или Бесцветно или
МПК / Метки
МПК: C07D 401/02, A61K 31/495
Метки: кислоты, фармацевтически, приемлемых, 6-фтор-1,4-дигидро-4-оксо-7-замещенной, пиперазинилхинолин-3-карбоновой, солей, получения, производных, способ
Код ссылки
<a href="https://kz.patents.su/7-1232-sposob-polucheniya-proizvodnyh-6-ftor-14-digidro-4-okso-7-zameshhennojj-piperazinilhinolin-3-karbonovojj-kisloty-ili-ih-farmacevticheski-priemlemyh-solejj.html" rel="bookmark" title="База патентов Казахстана">Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты или их фармацевтически приемлемых солей</a>
Способ получения замещенных аминодикислот, их рацематов или оптических изомеров, или их фармацевтически приемлемых солей

Номер патента: 1223
Опубликовано: 15.09.1994
Авторы: Жорж Ремон, Мишель Лоби, Мишель Винсен
МПК: C07D 217/26, A61K 31/47
Метки: замещенных, изомеров, способ, солей, приемлемых, оптических, рацематов, аминодикислот, фармацевтически, получения
Формула / Реферат:
Способ получения замещенных аминодикислот общей формулы 1 где А - бензольный цикл, n=1; А - насыщенный цикл n=0 или 1; R1 - низшая алкильная группа с 1-4 атомами углерода, содержащая аминогруппу; R2 - атом водорода или алкильная группа с 1-4 атомами углерода, R3 - линейная или разветвленная алкильная группа с 1-8 атомами углерода, моно- или ди- циклоалкил-алкильная группа или фенил-алкильная группа, содержащая в целом до 9 атомов углерода или...
Способ получения производных акриловой кислоты или их стереоизомеров

Номер патента: 1222
Опубликовано: 15.09.1994
Авторы: Ян Фергусон, Пол Дефрейн, Кристофер Ричард, Эйлз Годфри, Майкл Гордон Хичингс, Патрик Джелф Кроули, Вивьенн Маргарет Энтони, Джон Мартин Клаф
МПК: C07D 213/70, A01N 43/54
Метки: получения, кислоты, способ, стереоизомеров, акриловой, производных
Формула / Реферат:
Изобретение относится к гетероциклическим соединениям, в частности к получению производных акриловой кислоты ф-лы где W - пиридинил или пиримндинил, возможно замещенные галогенами C1-С4-алкилом, который, в свою очередь, может быть замещен галогеном, фенилом, С1-С4-алкоксигруппой; феноксигруппой, которая, в свою очередь, может быть замещена 1-метоксикарбонилом, 2-метоксиэтенилом, галогеном, циано- или нитрогруппой, амино-, циано-,...
Способ получения Е-изомеров производных акриловой кислоты

Номер патента: 1225
Опубликовано: 15.09.1994
Авторы: Патрик Джелф Кроули, Вивьенн Маргарет Энтони, Майкл Гордон Хичингс, Пол Дефрейн, Джон Мартин Клаф, Кристофер Ричард Эйлз Годфри, Ян Фергусон
МПК: A01N 43/54, C07D 239/30
Метки: получения, акриловой, кислоты, способ, производных, е-изомеров
Формула / Реферат:
(57) Изобретение относится к гетероциклическим соединениям и. в частности, к получению Е-изомеров производных акриловой кислоты формулы где W - пиридинил- или пиримидинилгруппа, замещенная галогеном, С1-С4-алкилом, который, в свою очередь, может быть замещен галогеном, фенилом, С1-С4-алкоксилом, феноксигруппой, которая может быть замещена 1-метоксикарбонил-2-метоксиэтенилом, галогеном, циано- или нитрогруппой, аминоформамидо-, нитро-, циано- или...
Способ получения 6 – метил – 3,4 – дигидро – 1 , 2 , 3 – оксатиазин 4 – он – 2 , 2 диоксида или его калиевой соли и способ получения аммоний ацетоацетамид – N – сульфонатов

Номер патента: 1228
Опубликовано: 15.09.1994
Авторы: Дитер Ройшлинг, Адольф Линкис, Карл Клаус
МПК: C07C 143/86, C07D 291/06, A23L 1/236...
Метки: оксатиазин, метил, калиевой, соли, сульфонатов, аммоний, получения, способ, диоксида, дигидро, ацетоацетамид
Формула / Реферат:
Изобретение касается гетероциклических соединений, в частности 6-метил-3,4-дигидро-1,2,3-оксатиазин-4-он-2,2-диоксида и его калиевой соли (ОТА), которые как обладающие сладким вкусом находят применение в пищевой промышленности, также новых промежуточных продуктов - аммоний ацетоацетомид-N-сульфонатов (ААС) формулы СН2-С(О)-СН2-С(О)-NH-SO3 [R1(R2)2R3N ], где R1-R2-R3 - Н, или R1 - Н, низший алкил; R2-низший алкил; R3-Н, низший алкил, фенил,...
Способ получения производных 3-окси-2-циклогексен-1-она

Номер патента: 1217
Опубликовано: 15.09.1994
Авторы: Винфрид Рихарц, Гернот Рейссенвебер
МПК: C07C 49/713
Метки: получения, производных, 3-окси-2-циклогексен-1-она, способ
Формула / Реферат:
Изобретение относится к циклическим кетонам, в частности к получению производных 3-окси-2-циклогексен-1-она формулы CH2-CHR1-CH2-C(O)-C = [С(О)R2] - С(ОН), где R1 - низший алкенил. С6 - циклоалкил, который может содержать одну олефиноненасыщенную связь, низший алкилтионизший-алкил, С7-12 -бициклоалкил, который может содержать две олефиноненасыщенные связи, фенил, незамещенный или замещенный низшим алкилом или галоидом, или низшей алкилтиогруппой...
Предыдущий патент: Способ получения 6 – метил – 3, 4 – дигидро – 1 , 2, 3 – оксатиазин – 4 – он – 2, 2- диоксида .
Следующий патент: Способ получения производных пиридина
Случайный патент: Устройство для быстрого охлаждения частиц расплава