Способ получения производных изоксазола







Формула / Реферат
Изобретение касается гетероциклических веществ, в частности получения производных изоксазола общей ф-лы I:
А-О(СН2)7-С=СН-С(СН3) =N-О,
где А - группа ф-лы II или III:
-C=CH-CH-CK-S (II); -C=CH-S-CHK-CH2> (III) при К --С=N-(СН2)2-O, которые обладают антивирусной активностью, что может быть использовано в медицине. Цель - создание более активных веществ указанного класса. Синтез ведут циклизацией соединений ф-лы:
IV или V: HO-(CH2)2-NH-C(O)-X1-O-(CH2)7-C=CH-C(CH3)=N-О (IV); HO-(CH2)2-NH-C(О)-X2-0-(CH2)7-C=CH-C(CH3)=N-О(V),
где X1 - и Х2
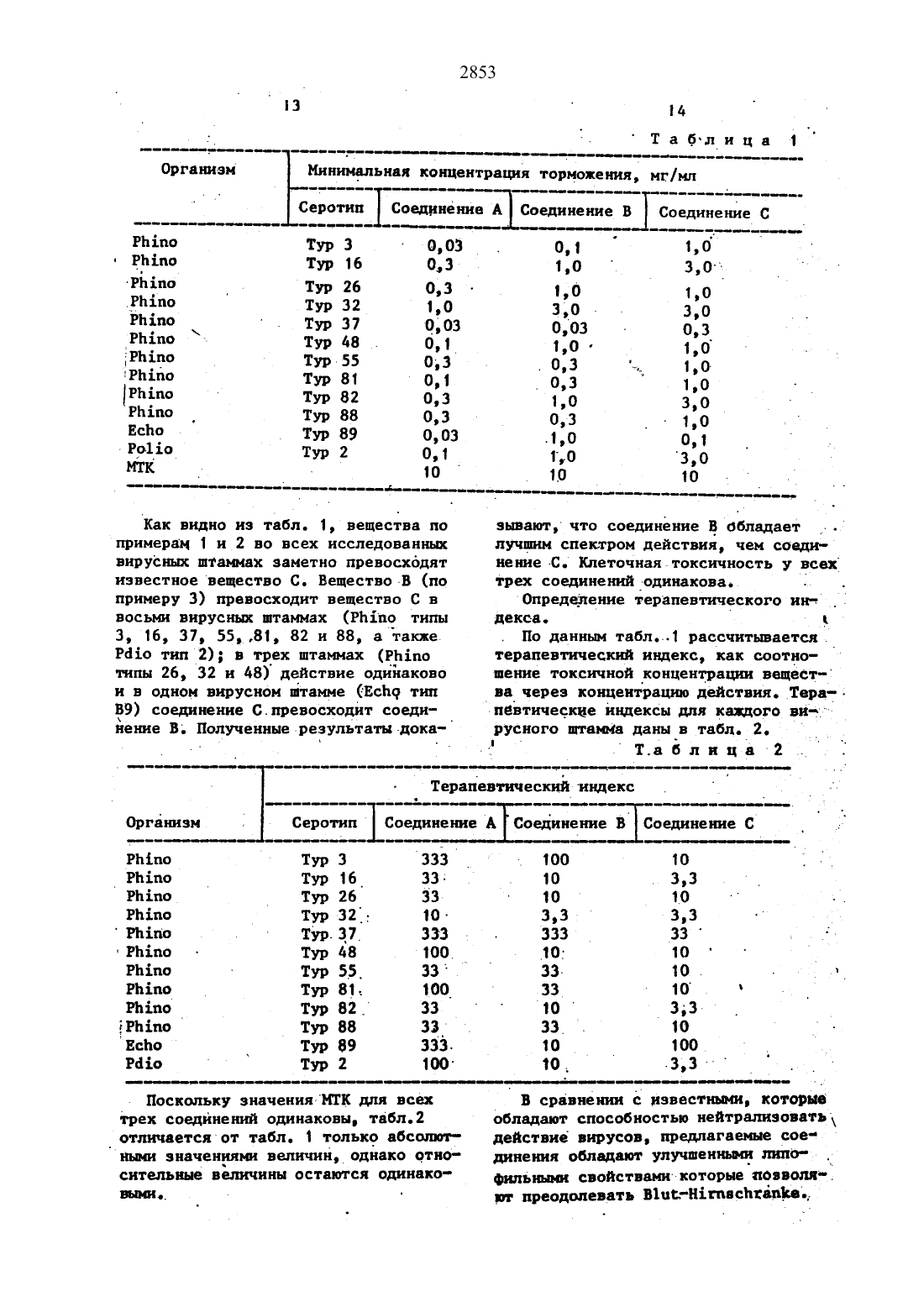
с помощью агента дегидратации и хлористого тионила в среде инертного растворителя. Новые соединения при антивирусной активности обладают лучшими в сравнении с известными липофильными свойствами, которые позволяют преодолевать Blut-Hirnschranke. 2 табл.
Текст
чество хлористого тионила. Остаток распределяют между насыщенным раствором кнслого углекислого натрия и. этиловым эфиром уксусной кислоты. Затем еще дваждыпроизводят экстрагирование этиловы эфиром уксусной кислоты. Объединенные органческие растворы сушат над сернокислым натрием при добавлении активированного угля,а эатемупаривают.неочищенный продукт (0,52 г окрашенного вжелтоватый цвет кристаллического вещества) очищают с помощью хроматографии на колонке (135,.силикагель 60, раэмер зерен 0,04 О 0,063 элюирующее средство этиловый эфир уксусной кислоты и петролейный эфир в соотношении 31). .Выод 0,29 г бесцветного кристаллического вещества (50,71 от теоретически рассчитанного значения). Т.пл. б 970 С (из диизопропилового эфира). . , Исходый материал получают описанным ниже способом.21,0 г (0,216 моль) 3,5-диметилизоксазола растворяют в 200 мл абсолютного тетрагидрофурана,.раствор охлаждают до 80 С, после чего при указанной температуре к раствору в течение 40 мин прибавляют по каплям 160 мл раствора нбутиллнтия (135 М раствор в н-гексане, 0,216 моль). Затем смесь дополнительно перемешивают в течение 15 мни при -75 С.Непосредственно после этого реакционную смесъ прибавляют по каплям к раствору 53,5 г (0,217 моль) 1 нод 6 хлоргексана в 15 О мл абсолютного ж тетрагидрофурана таким образом, чтобы температура не поднималась выше 60 С. После завершения прибавления реакционную смесь дополнительно перемешвают в течение 15 мин при-6 ОС а затем температуре смеси дают воэмюжность подняться до комнатной температуры.Реакционную смесь распределяют между хлористым метиленом и 0,2 и.раствором соляной кислоты, водную фазу еще три раза экстрагируют хлористым метиленом, объединенные органические фазы сушат над сернокислым натрием и упаривают.Выход 26,92, окрашенное в желтоватый цвет маслообразное вещество.(47,4 ммоль) иодида натрия в течение 24 ч нагревают в 60 мл абсоптного ацетона при температуре кипения реак ционной смеси с обратным холодильни- дком. Непосредственно после этого реакционную смесь охлаждают, выделившийся в осадок хлористый натрий отфильтровышают, промывают небольшим количеством ацетона,.после чего фильтрат смешивают с 7,16 г(94,8 моль) углекислого калия. Смесь нагревают с обратным холодильником при темературе ее кипения в течение2 ч, охлаждают и затем упаривают. Остаток распределяют между водой и диэтиповым эфиром, после чего водную . фазу дважды экстрагируют диэтиловым эфиром. Объединенные органические растворы промывают небольшим количеством насыщенного раствора гидро сульфита натрия, сушат над сернокис ЛЫН НЗТРИЗМ при добавлении 8 КТИВИР 0ванного угля, ааатем упаривают. . неочищенный продукт (11,9 г 821 от теоретически рассчитанного значения) перекристаллизовывают из димэо-1 пропилового эфира). Вымод 5,2 г окрашенного в светлод 1РОЗОВЫЙ ЦВЕТ КРИСТЗЛЛНЧВСКОГО ВЕЩВСТ0,98 г (2,90 ммоль) метилового эфира 57-(3 метил 5 изоксазолил) гептилокси 2 тиофенкарбоновой кислоты нагревают в.8 мл этилового спирта-и 4 мл воды с обратным холодильником до температуры кипения смеси, после чего.к полученному раствору в течение 10 мин прибавляют по каплям 0,18 г (319 ммоль) гидроокиси калия,растворенной в 6 мл воды и 4 мл этилового спирта. Непосредственно после этого реакционную смесь дополнительно перемешивают в течение 2,5 ч с обратны холодильником при температуре ее кипения. , После охлаждения большую часть смеси отгоняют, остаток распределяют между водой и диэтиловым эфиром, а водную фазу затем подкисляют.прибав ленем 2 н. соляной кислоты до рН 1,5. Затем применяют 80 мл последнего, объеднненные органические раство 4,04 (С, РЫ СУШЗТ над СВРНОКИСЛЬЩ натрием придобавлении ЗКТИВНРОВЗННОГО УГЛЯ И.лического вещества (95,82 от теоре тически рассчитанного значения). Т.пл. 96-97 С (из диизопропилового эфира). .К 0,81 г (2,51 ммоль) 57(3 метилч 5 изоксазолнл)-гептилоксич 2-тио фениарбоновой кислоты при леремешивани и охлажденн медленно прибавляют по каплям 2 мл хлористого тионила, в результате чего образовывается проарачыираствор. Затем реакционную -смесь дополнительно перемешвают в течение 30 мин при комнатной темпера 6туреи непосредственнопосле этоо производят отгонку в вакууме избыточного количества хлористого тионила. Остаток растворяют в 6 мл абсолютного хлористого метилена, после чего при 15 С к приготовленному раствору прибавляют по каплям раствор 0,34 глютного хлористого метилена. Реакционную смесь дополнительно перемешивают 5 течение 1 ч припкомнатиой температуре, несколько упаривают и затем распределяют между водой и этиГ ловым эфиром уксусной кислоты. водную фазу еще раз экстрагируют небольшим количеством этилового эфира уксусной. кислоты, объединенные органические растворы промывают водой, сушат над сернокислым натрием при добавлении активированного угля и упариваюттВыиод 0,81 г окрашенного в желтоватй цвет кристаллического вещества (88,22 от теоретически рассчитанного значения). Т. пл. 107110 С-гептилокси 2 тиофенкарбоновой кис. лоты растворяют в 30 мл хлороформа,после чего приготовленны раствор смешивают при 0 С с 0,3 г(2,5 д ммоль) хлористого тионила. Непосредственно после этого реакционную смесь перемешивают в теченелучеют сухой остаток. Остаток распре 6 деляют между иасыениым раствором кксглого углекислого натрия и этиловмэфиром уксусной кислоты. Еще дна ра за производятэкстрагирование зтиповы эфиром уксусной кислоты, объеди- неннне органические растворы сушатнад сериокнслым натрием при добавлеи активированного угля и упаришают.ОЧНСТКУ, НЗОЧЪЩЕННОГО ПРОДУДСЪЗ С -. ЛОПЮЦЬЮ ХРОНДТОГРЗФНН на КОЛОНКЕ ОСУшествляют по аналогии с описанным в приере 1. .Выход 0,24 г бесцветного кристаллического вещества (422 от теоретич чески рассчитанного значения). Т.пл. 68-70 С (из диизопропилового эфира).2,06 г (5,б 2 ммоль) Ы-(2-оксиэтил) амид-4-С 7-(3 метил-5-изоксазолил)гептипокси-2-тиофенкарбоновойкислоты при 0 С вводят в 5 мл хлористого тионила.Смесь перемешивают в тече- ние 10 мин при 0 С, после чего в вакууме без нагревания удаляют избыточное количество хлористого тионила. Остаток распределяют между насыщенным раствором кислого углекислого натрия и этиловым эфиром уксусной кислоты. Еще два раза производят экстрагирование этиловым эфиром-уксусной кислоты, объединенные органические растворы сушат над сернокислы натрием при добавлении активированного угля и упаривают. Неочищенное маслообразное вещество подвергают очистке с помощью хроматографии на колонке (140, силикагель 60, размер зерен от 0,040 до 0,063 элюирующие средст-во этиловый эфир уксусной кислоты-иВыиод 0,52 г окрашенного в желтоватый цвет кристаллического вещества(26,52 от теоретически рассчитанного значения). Т. пл. 67-б 8 С (из диизопропилового эфира). уИсходный материал получают описанным ниже способом.кипения, после чего к приготовленной смеси прибавляют по каплям в течене 20 мин 43,7 г (0,347 моль) диметил сульфата. Затем реакционную смесь до 8волнительно нагревают в течение.2,5 ч с обратным холодильником притемературе кипения. Непосредственно после этого реакционную смесь упарнвают в вакууме, остаток распределяют между насыщенным раствором углекислого натрия и диэтиловым эфиром, водную фазу еще пять раз экстрагируют диэтиловым эфиром, причем каждый раз применяют по 80 мл последнего. Объединенные органические растворы сушат над сернокислым натрием при добавлении активированного угля, фильтруют и упарива ют. Выщод 49,6 окрашенного в желтоватьй цвет кристаллического веществазначения). Т. пл. 84-85 С (из смеси динзопропилового эфира и петролейного эфира). ГныМк (свс 1,) ю ррт 7,22 (а,д 1,7 Н 2 1 Н, ТЬ-Н 3), 6,64 (д, 5 17 на, 1 Н, ТЬ-Н 4) 6,70 (5, 1 Н,он) 3,88 (5, 3 н,осн,). 5-(7-Иодгептил)-Зметилизоксазол. 16,66 г (77,23 ммоль) 5(7 хлор гептил)-3-метилизоксазола и 12,75 г(85,06 ммоль) иодистого натрия нагревают с обратным холодильником в 110 мл безводного ацетона при температуре кипення смеси.По данным Н-ЯМтспектроскопии че рез 7 ч степень превышения составляет приблизительно 852, а через 22 чприблизителвно 892. Через 27 ч реак. цнонную смесь упаривают и остаток.распределяют между-дилорметаном и 6 водой (при добавлении нескольким иллилитров 2 н. соляной кислоты. водную фазу несколько раз экстрагируют дн-х хлорметаном причем суммарно применяют 250 мл последнего, и органическую фазу сушат над сернокислым натрием и упаривают. .Выход 22,95 г окрашенной в коричневы цвет жидкости (96,72) от теоретически рассчитанного значения).(4060 ммоль)-углекислого калия в 130 мл безводного ацетона при тем пературе КИПЕНИЯ СМЕСИ С обратим ХОлодильииком. Реакционную смесь вндерживают в течение ночи, а затем упаривают. Остаток распределяют между 2 Но Растворомгидроокиси натрия и диэтиловым эфиром, водную фазу ещевым эфиром, причем сумарно применяют 150 мл-последиегот органическую фазу сушат над сернокислым натрием при добавлении активированного угля и упаривают. чцвет кристаллического вещества (9532от теоретически рассчитанного значе них).11,34 г (3361 ммоль) метилового эфира 4-Г 7-(3 метил 5 изоксазолил)гептилокси 3-2-тиофенкарбоновой кислоты нагревают с обратным холодильнком в 95 мл этилового спирта и 45 млВОДЫ ДО ТЗМПЕРЗТУРЫ кипения, ПОСЛЕчего к приготовленному раствору прибавляют по каплям 2,18 г (38,9 ммоль) гидроокиси калия, растворенного вПосле нагревания в течение 3 ч при температуре кипения с обратным холодильником реакционную смесь охлаждают, упаривают остаток распределяют между водой и диэтиловым эфиром, а водную фазу после подкисления 2 н. соляной кислотой до рН 1 еще НеСК 0 ЛЬко раз экстрагируют диэтиловымэфи ром. Объединенные органические растворы сушат над сернокисльм натрием при добавлении активированного УГдЯцвет кристаллического вещества (85,61Неочищенный продуктнепосредствен но может быть применен на последуюцейстадии или может быть перекристаллнзоваи из диизопропилового эфира, в результате чего образуется бесцветное кристаллическое вещество.фенкарбоиовой кислоты при 0 С прибавляют приблиэительно 15 мл хлористого тионила, после чего реакционную смесь в течение 20 мин перемешивают при комнатной температуре. Полученный после фильтрования с применением вакуума водоструйного насоса остаток растворяют в д 0 мл безводного дилорг метана, после чего к приготовленному раствору при охлаждении прибавляют по каплям раствор 2,2 г (3609 ммоль) этаноламмна в 40 мл безводного ди-мы хлорметана. Реакционную смесь перемешивают в течение 2 ч прикомнатной температуре, затем упаривают остаток распределяют между водой и дихлорметаиом, причем для улучшения раэделения.фаз прибавляют небольшое количество 2 н. раствора соляной кислоты. Полученную после разделеня А фаз органическую фазу сушат над сернокнслы натрием при добавлении активированного угля и упаривают. Выход 5,74 г окрашенного в коричневый цвет кристаллического вещества(95,6 от теоретически рассчитанного значения) Т.пл. 56-57 С.(неочищенный продукт без уменьшения выхода непосредственно может быть примененДЛЯ ПОЛУЧСНИЯ СОЕДИНСННЯ предлагаемой
МПК / Метки
МПК: C07D 413/14
Метки: производных, способ, изоксазола, получения
Код ссылки
<a href="https://kz.patents.su/8-2853-sposob-polucheniya-proizvodnyh-izoksazola.html" rel="bookmark" title="База патентов Казахстана">Способ получения производных изоксазола</a>
Способ получения производных 1, 2, 5, 6-тетрагидропиридин-3-карбоксальдегидоксима или их гидрохлоридов

Номер патента: 1858
Опубликовано: 15.03.1995
Авторы: Алина Бутти, Фернандо Бардзаги, Джулио Галлиани, Карла Бонетти, Эмилио Тоя
МПК: C07D 211/70, A61K 31/44
Метки: получения, гидрохлоридов, 6-тетрагидропиридин-3-карбоксальдегидоксима, производных, способ
Формула / Реферат:
Изобретение относится к гетероциклическим соединениям, в частности к получению производных 1,2,5,6-тетра-гидропиридин-3-карбоксальдегидроксима ф-лы где R1-Н или метил, R2-линейный или разветвленный насыщенный С1-3-алкил, линейный или разветвленный ненасыщенный C2-4 -алкил, ацетил или диметиламиноэтил, или их гидрохлоридов, обладающих холиномиметической -активностью. Цель изобретения - выявление новых более активных соединений указанного класса....
Способ получения производных 2-тиенилоксиуксусной кислоты или их фармацевтически приемлемых солей

Номер патента: 1567
Опубликовано: 15.12.1994
Авторы: Дитер Биндер, Франц Ровенсцки, Хуберт Петер Фербер
МПК: A61K 31/38, C07D 333/26
Метки: производных, кислоты, приемлемых, 2-тиенилоксиуксусной, способ, фармацевтически, солей, получения
Формула / Реферат:
Изобретение касается производных 2-тиенилоксиуксусной кислоты, в частности получения 5-[2-(бензолсульфониламино)этил]- или 5-[2-(4-хлорбензолсульфониламино) этил]-2-тиенилоксиуксусных кислот, которые могут быть использованы в медицине для лечения тромботических заболевании. Цель - создание новых более активных веществ указанного класса. Синтез ведут окислением, например, амида N-[2-[2-(5-(2-гидрокси)-этокси)-тиенил]-этил]-бензолсульфоновой...
Способ получения производных акриловой кислоты или их стереоизомеров

Номер патента: 1222
Опубликовано: 15.09.1994
Авторы: Майкл Гордон Хичингс, Ян Фергусон, Джон Мартин Клаф, Патрик Джелф Кроули, Кристофер Ричард, Вивьенн Маргарет Энтони, Пол Дефрейн, Эйлз Годфри
МПК: C07D 213/70, A01N 43/54
Метки: получения, акриловой, способ, кислоты, производных, стереоизомеров
Формула / Реферат:
Изобретение относится к гетероциклическим соединениям, в частности к получению производных акриловой кислоты ф-лы где W - пиридинил или пиримндинил, возможно замещенные галогенами C1-С4-алкилом, который, в свою очередь, может быть замещен галогеном, фенилом, С1-С4-алкоксигруппой; феноксигруппой, которая, в свою очередь, может быть замещена 1-метоксикарбонилом, 2-метоксиэтенилом, галогеном, циано- или нитрогруппой, амино-, циано-,...
Способ получения производных цефалоспорина или их легко гидролизуемых сложных эфиров, или их солей с щелочными металлами

Номер патента: 1574
Опубликовано: 15.12.1994
Авторы: Рене Эймэ, Дидье Пронин
МПК: C07D 501/06, A61K 31/545
Метки: сложных, цефалоспорина, легко, гидролизуемых, эфиров, способ, получения, солей, металлами, производных, щелочными
Формула / Реферат:
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЦЕФАЛОСПОРИНА ИЛИ ИХ ЛЕГКО ГИДРОЛИЗУЕМЫХ СЛОЖНЫХ ЭФИРОВ ИЛИ ИХ СОЛЕЙ С ЩЕЛОЧНЫМИ МЕТАЛЛАМИ формулы I в виде син-изомеров, где R1 - атом водорода, С1-С2 алкил, возможно замещенный атомом брома, карбоксиметил, этоксикарбонил, бензил, бензоил, 1-оксигексадецил или циклододецилоксикарбонил, R2 - атом водорода, остаток легко гидролизуемой сложно-эфирной группы или катион щелочного металла, такого как натрий, R3 - прямой...
Способ получения производных плевромутилина или их гидрохлоридов

Номер патента: 1560
Опубликовано: 15.12.1994
Авторы: Херманн Выплель, Хайнц Бернер
МПК: A61K 31/10
Метки: получения, плевромутилина, производных, способ, гидрохлоридов
Формула / Реферат:
Изобретение относится к полициклическим соединениям, в частности к получению производных плевромутилина ф-лы где R1-C1-С6-аминоалкил, или их гидрохлоридов, которые проявляют антимикробную активность. Цель - получение более активных соединений. Получение ведут реакцией соответствующего замененного амина со сложным эфиром соединения ф-лы HOOCR2, где R2- С1-С6-алкиламин, аминогруппа которого содержит защитные группы (такую как С(O)ОС(СН3)3) при...
Предыдущий патент: Способ получения производных простых арилфениловых эфиров или их кислотно-аддитивных солей, или их металлических комплексов.
Следующий патент: Способ получения диазабицикло-(3, 3, 1)-нонанов
Случайный патент: Способ изготовления поковок