Способ получения производных 2-тиенилоксиуксусной кислоты или их фармацевтически приемлемых солей
Номер патента: 1567
Опубликовано: 15.12.1994
Авторы: Дитер Биндер, Франц Ровенсцки, Хуберт Петер Фербер





Формула / Реферат
Изобретение касается производных 2-тиенилоксиуксусной кислоты, в частности получения 5-[2-(бензолсульфониламино)этил]- или 5-[2-(4-хлорбензолсульфониламино) этил]-2-тиенилоксиуксусных кислот, которые могут быть использованы в медицине для лечения тромботических заболевании. Цель - создание новых более активных веществ указанного класса. Синтез ведут окислением, например, амида N-[2-[2-(5-(2-гидрокси)-этокси)-тиенил]-этил]-бензолсульфоновой кислоты окисью серебра в водно-щелочной среде. Растворяют амид в 0,5-4 н. преимущественно в 2 н., растворе КОН или NaOH, окисляют 2-16 эквивалентами окиси серебра при 75-85°С в течение 2-3 ч. Исходную окись серебра предварительно получают растворением AgNO3 в дистиллированной воде с добавлением эквимолярных количеств NaOH. Целевой продукт выделяют в свободном виде или в виде фармацевтически приемлемой соли. Новые соединения малотоксичны и в 4 раза активнее, чем дазоксибен, при этом начало антитромбического действия наблюдается при 1 мг/кг и достигает максимума при 5 мг/кг против 20 мг/кг.
Текст
тровывают и проишают многократно дистиллированной водой.(5-(2 оксн)-этокси)7 тиеннлТ-этилЧ бензолсульфокислоты (1) растворяют в 90 мл водного раствора гидроокиси натрия,-добавляют еще влажную окись серебра ипрнмеканич 2 скОМ ПЕРЕМЭШИ ваиии нагревают до 80 С. Спустя 3 ч при этой температуре охлацдают н суспензию фильтруют через,НТЕЬОПроэрачньй раствор гидроокиси натрияподкисляют с помощью примерно 6 мл,концентрированной соляной кислоты и экстрагируют триды по 100 мл эфира. Эфирные фены встряхивают трижды по100 мл с насыщенным раствором бикар боната натрия, промывают их 50 мл зфнраи подкисляют.концентрированной соляной кислотой. водную фаэу.экстратируют дважды с-помощью 150 мл эфире, объединенные эфирные фазы сушат над сульфатом натрия, отфильтровывают И выпаривают. КРНСТаЛЛНЧеСКИЙосадок-настаивают с 30 мл диизопропи лового эфира и отфильтровывают. Еьтод 2,6 г бесцветных кристаллов(трнплет, О-СНд 7 С 0) 43,8 (трнплет,ННСНд) 30,2-(трнплет ТЬ-СН-). бискодный продукт можно получать следующим обравОн 2(2-Тиенилокси)этанол.К 1600 мл абсоютиого этиленглн коли добавляют 323,1 ми 5,4 Н раствора метнпата натрия(175 моль). Реакцит онную смесь нагревают н 0 бРаЭУЮЩИЙсЯМЕТЕНОП ОТГОНЗЮТ ПУТМ пропусканииазота через-обратный холодильник до тех пор, пока температураавниней части не повысится до 130 С. По окон 4 чании удаления метанола добавляют 187,5 г (1,15 моль) 2 бромтиофена,- 55,5 гтонко измельченной окиси ме ди и 5,6 г иодида натрия, аппаратуру еще короткое-время промывают азотом,закрывают баллон и перемешивают 175 ч при 80 С. Затем реакционную смесь охлаждают н отсасывают через НУЕЬ 0. Фильтрат разбавляют 800 мл воды и слегка иодкисляют концентрированной соляной кислотойЗкстрагируют четыре раза по 400 мл метиленхлоридом (в целом 1600 мл). Объединенные органические фазы встряхивают один раз с 200 мл воды, сушат надсульфатом натрия, отфильтровывают И выпариваюто Остаток перегоняют.Охлаждают до 20 С и при переменгивании прикалывают 83,2 мл.(02 О 8 моль) -2,5 М раствора нбутил лтияв Н-гексане так, чтобы температура не превышала 10 С. Оставлнют нагреваться до комнатной температуры и перемешвают в течение часа. О Реакционную смесь охлаждают до 10 С н в течение 30 мин прикалывают раствор 19,О 5.г (0,1 Од моль) Нбензолсульфонилавнрндина в 100 мл абсо лютного ТГФ при 10-15 С. Все нагревается до комнатной температуры и перемешвается еще 2 ч. Смесь выливают на 200 мл 2 н. вод ного раствора.НС 1 и экстрагируют трижды по 120 мл метипенхлоридом. Объединенные органические фазы сушат над сульфатомнатрия, отфипьтровывают н выпаривают 0 статок обрабатывад ют 300 мл абсолютного метаноласме шивают с 2 мл 302 ной.метанопьной соляной кислоты и перемешивают 10 мин при комнатной темературе. Последобавки 2 г карбонатанатрия вьшаривают в вакууме.Остаток распределяют между 250.мп 1 н. водного раствора гидроксиланатрия и 200 мл эфира, и эфирную фазу допопнительн 0 промЫвают одинразпроывают дважды по 10 мл эфиром,подкисляют примерно 25 мл концентрированной соляной кислоты и экстрагируют трехкратно, смотря пообстоя тельствам 150 мл метиленхлорида. Метиленхлоридную фазу.СУШаТ НЭДСУПЬФЭ том натрия, смешивают с Зрг активного угля, отфилътровывают и выпаривают. Выход 33,3 г темно-красного масла (9782 от теории), которое используется непосредственно на самой блиайшей стадии.П р и м е р 2. Растворяют г 5-2-(бензолсупьфониламино)-зтил 2 тиенилоксиуксусдой кдТЫ (293 ММ 0 ЛЫ в 20 мл метанола и прикалывают 0,117 г На 0 Н (293 ммоль), раствореннш в 10 мл метанола, и перемешивают раствор в течение 1 ч при коматиой температуре. В заключение упарнвают досуха, остаток промывают этилацетатом и диизопропиловым эфиром и сушат в вакууме при 50 С. цРастворяют четыре порции по 9,3 г(0,02 В моль) амида Н-2-(5(2 гидрокси)-этокси)-тненшл-этил-бензолсульфоиовой кислоты (11) в 90 мл 2 Н ВОДНОЙ натриевой щелочи каждая добавляют еще влажную окись серебра и нагревают при перемешивании додалънейщанобработка происходит как в примере 1. Все данные сведены в табл. 176 П р и м е р й. 5-2-(4-Хлорбензолсульфониламшно)этил-2-тиенилок СИУКСУСНЗП КИСЛО Та .15 г (0,088 моль) нитрата серебра растворяют в 90 мл дистиллированной воды и при перемешивании медленно прикалывают раствор 35 г(0,088 моль)-гидроокиси натрия в 5 мл дистиллированной воды. Образо вавшуюся суспензию окиси серебра перемешивают еще 10 мин, осадок отфиль тровьшают и промывают многократно дистиллированной водой.4,0 г (0,011 моль) амида-4-хлорЫ--2-(5-(2-окиси)-этокси)тиенил этилбензолсульфокислоты(11) растворяют в 40 мл 2 н. водного раствора гидроокиси-натрия, добавляют еще влажную окись серебра И нагревают до 8 ОС при механическом перемешивании.Спустя 3,5 ч при этой температуре охлаждают и суспензию отсасывают через НЧРЪ 0 и дополнительно промывают 2 нводным раствором гидроокиси натрия. Прозрачный раствор гидроокиси натрия подкисляют с помощью концентрированной соляной кислоты и экстрагируют триды по 80 мл эфиром.Эфирные фазы дважды встряхивают сц насьщенным раствором бикарбоната натрия, отбирая кажды раз по 50 мл,промывают один раз 50 мл эфира и подкисляют концентрированной соляной кислотой. Водную фазу экстрагируют дважды по 150 мл эфиром, объединенные эфирные фазы сушат над сульфатом иатрии, отфнльтровывают и выпаривают,Сырой продукт перекристаллизуют из толуола. Въщод 1,3 г бесдветных кристаллов (31, от теории), т.пл 125127 С (толуол).Исходный продукт может быть полу 5 чен следующим образом18,9 г (0,137 моль) 2(27 тиенилокси)тэтанола помещает в 200 мл абсолютного тетрагидрофурана и растворяют тампримерно-50 мг.птолУ 0 лсульфокислоты. Добавляют 1 д,2-г(0169 моль).3,4-дигидропирана н перемешивают 8 Чы Охлаждают до 209 С и при перемешваиии прикалывают 66 мл (0165 моль) 2,5 М растворанбутиллития,в нгексане так, чтобы температура не превышала 15 С.нагревают до комнатной температурыРеакционную смесь выливают натрижды по 250 мл.метиленклоридом. Объединенные органические фазы сушат над сульфатом натрия, отфильтровывают и выпаривают. Остаток обрабатывают 100 мл абсолютного метанола, смеши.вают с.10 мл 3 Оной метанольной соля ной кислоты н перемешивают 10 минпри комнатной температуре. Добавляют одну ложку карбоната натрия и метанол отгоняют. Остаток распределяют между 100 мл 1 н. водного раствора гидроксида натрия н 100 мл эфира и эфирную фазудополнительно промшают один раз 100 мл 1 н.раствора гидроокиси натрия, объедишеныеуводные фазы промывают дважды.по 50 мл эфиром,подкислнют концентрированной соляной кислотой и экстрагируют трижды, смотря по обстоятельствам,.100 млметиленхлорида. Затем сушат надчсульфатом натрия, смешивают с активным углем, отфильтровывают и выпаривают. Попу чают 10,15 г вязкого темного масла. Этот сильно загрязненный сырой-проч дукт фильтруют через силикагель 60(153 от теории), тл. 8587 С (банд зол). Исследование антитромботической 1 активности. , самцов крыс Н 15 Еат(5 РР) массой 200-300 г.иаркотизируют с помощью пентобарбиталнатрин (60 мг/кг интраперитонеально).После этого животным внутривенноинъекЦируют вещество примера 1 (вещество А) дазоксибен т гидроклорид д 2 т(1 Нимидазол 1 ип)этокси-бензойной кислоты (вещество н ВВенолубрышейки) Чоответственно. Высвобождают препарированием, укрепляют в скобках на объективемикроскопа и споласкивают с.постояннойскоростЬю 2,5 мл/мин физиологическим раствором хлорида натрИн.Пазерныйлуч когерентности СЕ.2 суперграфит ного ионного лазера (аргоновы лазер) в течение 30 мин после инъекции испыг ТУЕМОГО еЩЕСТВа НЗПРНВЛЯЮТ ЧЕРЕЗ интерференционно-коитрастиыйобъектив БОХ микроскопа Ъе 1 Е 2 .0111 ор 1 ап с продолительностью 130 с на венолу Ислодная энергия ниже объективамикроскопа составляет 0,18 Вт.Если после первого лазерного пуска не образуетесли тромб по.длине и ширине не соответствует диаметру сосуда, то проИ 3 Б 0 дятвторой запуск лазера, чтобы получить тромб, который по длине и дгирине соответствует диаметру сосучда. .ЧИСЛО лазерных Пусков при этом является мерой антитромботической активности испытуемых веществ.чем больше число лазерных пусков при одинаковом диаметре сосуда, тем сильнее анти тромботический эффектВ качестве контролей служат животные, которые не получили никаких испытуемым веществд 1-Тесты осуществляются лоиспытуемому веществу и концентрации на 5 животных, причем животного повреждаются З-сосуда диаметром 2030.м. Статнстическаноценка осуществляетсявишшеп тесту по Пипп Результаты опытов представлены вмассы тела н достигает максимума дейч ствия при 5 мг/кгДальнейшее увелиЧЕННЕ Концентрации не приводит ни к
МПК / Метки
МПК: A61K 31/38, C07D 333/26
Метки: кислоты, фармацевтически, солей, 2-тиенилоксиуксусной, приемлемых, получения, производных, способ
Код ссылки
<a href="https://kz.patents.su/8-1567-sposob-polucheniya-proizvodnyh-2-tieniloksiuksusnojj-kisloty-ili-ih-farmacevticheski-priemlemyh-solejj.html" rel="bookmark" title="База патентов Казахстана">Способ получения производных 2-тиенилоксиуксусной кислоты или их фармацевтически приемлемых солей</a>
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты или их фармацевтически приемлемых солей
Номер патента: 1232
Опубликовано: 15.09.1994
Авторы: Ясуо Итох, Томио Сузуки, Эйити Косинака, Хидео Като, Нориюки Яги, Нобуо Огава
МПК: C07D 401/02, A61K 31/495
Метки: фармацевтически, пиперазинилхинолин-3-карбоновой, способ, кислоты, 6-фтор-1,4-дигидро-4-оксо-7-замещенной, приемлемых, солей, получения, производных
Формула / Реферат:
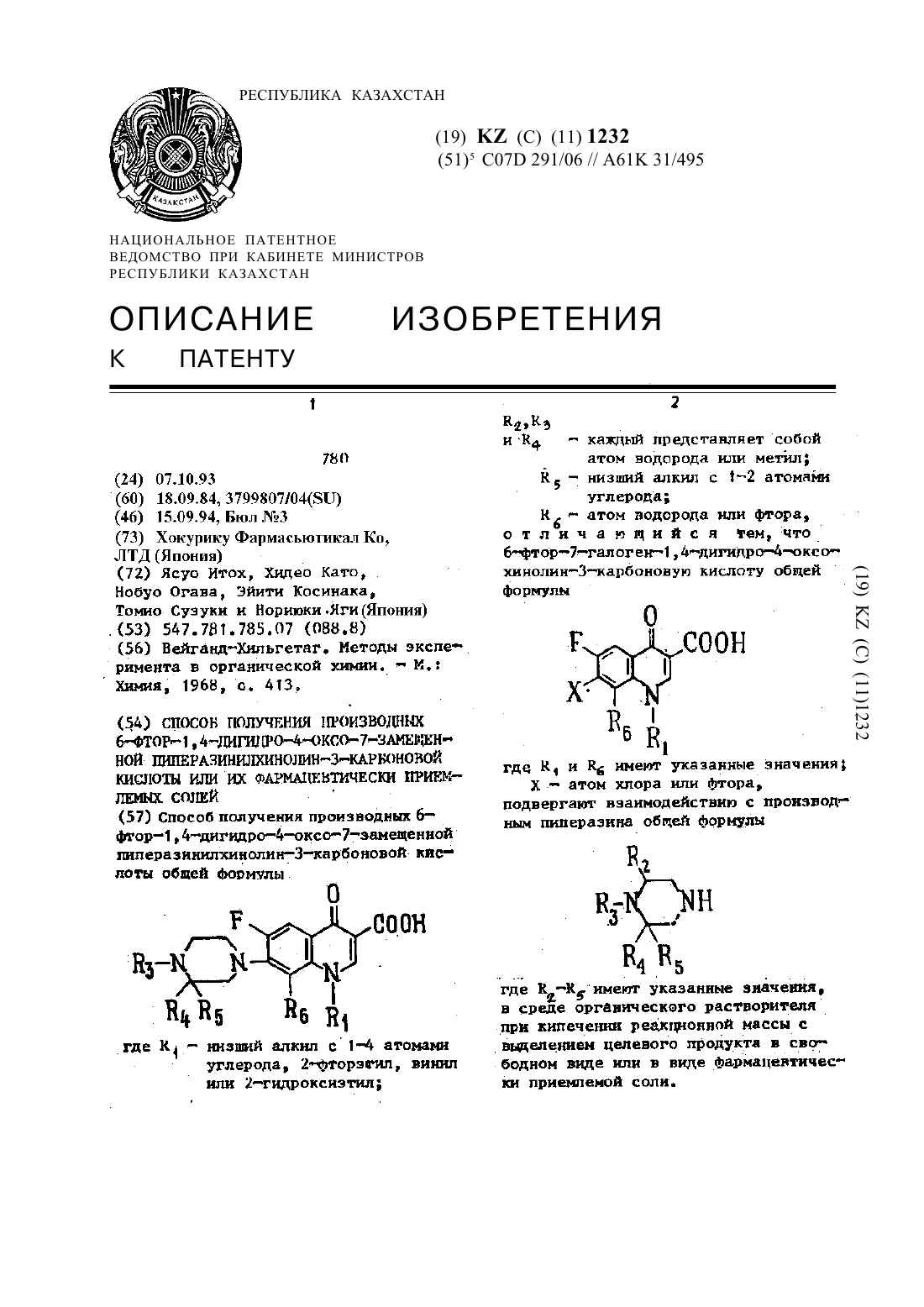
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты общей формулыгде R1 - низший алкил с 1-4 атомами углерода, 2-фторэnил, винил или 2-гидроксиэтил; R2, R3 и R4 - каждый представляет собой атом водорода или метил; R5 - низший алкил с 1-2 атомами углерода; R6 - атом водорода или фтора, отличающийся тем, что 6-фтор-7-галоген-1,4-дигидро-4-оксо-хинолин-3-карбоновую кислоту общей формулыгде R1...
Способ получения правовращающих производных оксиизоиндолинила или их фармацевтически приемлемых солей

Номер патента: 1561
Опубликовано: 15.12.1994
Авторы: Пьерлуиджи Гриджи, Джорджио Черони, Фабрицио Орци, Бруно Миорини, Джованни Карниель
МПК: C07D 209/46
Метки: способ, приемлемых, получения, оксиизоиндолинила, солей, фармацевтически, производных, правовращающих
Формула / Реферат:
Изобретение касается гетероциклических веществ, в частности правовращающих производных оксииэоиндолина общей формулы I где Х = C6H4-пaрa-CHR-C(O)-O-R1 ; R=C1-С4-алкил; R=H или С1-С4-алкил, или их фармацевтически приемлемых солей, обладающих обезболивающей и противовоспалительной активностью, что может быть использовано в медицине. Цель изобретения - повышение выхода целевого продукта. Синтез ведут реакцией офталевого ангидрида с правовращающим...
Способ получения замещенных аминодикислот, их рацематов или оптических изомеров, или их фармацевтически приемлемых солей

Номер патента: 1223
Опубликовано: 15.09.1994
Авторы: Мишель Лоби, Мишель Винсен, Жорж Ремон
МПК: A61K 31/47, C07D 217/26
Метки: способ, солей, аминодикислот, оптических, фармацевтически, замещенных, приемлемых, рацематов, изомеров, получения
Формула / Реферат:
Способ получения замещенных аминодикислот общей формулы 1 где А - бензольный цикл, n=1; А - насыщенный цикл n=0 или 1; R1 - низшая алкильная группа с 1-4 атомами углерода, содержащая аминогруппу; R2 - атом водорода или алкильная группа с 1-4 атомами углерода, R3 - линейная или разветвленная алкильная группа с 1-8 атомами углерода, моно- или ди- циклоалкил-алкильная группа или фенил-алкильная группа, содержащая в целом до 9 атомов углерода или...
Способ получения замещенных 1-(1-фенилциклобутил )- алкиламинов или их фармакологически приемлемых солей

Номер патента: 1554
Опубликовано: 15.12.1994
Авторы: Эрик Чарльз Вилмшерст, Джеймс Эдвард Джеффри, Антонин Козлик
МПК: C07C 87/34, A61K 31/135
Метки: 1-(1-фенилциклобутил, приемлемых, солей, замещенных, фармакологически, алкиламинов, способ, получения
Формула / Реферат:
Изобретение относится к амидам карбоновых кислот, в частности к получению замещенных 1-(1-фенилциклобутил)-алкиламинов общей формулы (I) где, если n=0,то R1-С1-С6-алкил, С3-С7-циклоалкил, циклогексилметил, 3-бутенил, фенил, фторфенил или метоксифенил и если n=1, то R1-Н, R2 - Н, метил или формил, R3 и R4 (независимо) - Н или галоген, трифторметил, метил, иэопропил, метоксигруппа или фенил, или совместно с соседними атомами углерода образуют...
Способ получения эфиров – производных циклопропанкарбоновой кислоты

Номер патента: 1552
Опубликовано: 15.12.1994
Авторы: Жан Тессье, Жак Мартель, Андре Теш
МПК: A01N 53/00, C07C 69/743
Метки: кислоты, получения, циклопропанкарбоновой, производных, способ, эфиров
Формула / Реферат:
Изобретение касается производных циклоалифатическнх кислот, в частности получения эфиров замещенной циклолропанкарбоновой кислоты общей формулы R-O-C(O)-CH=CH-СН-С(СН3)2-СН-С(О)OА1 , где R-C1-C4-алкил, замещенный хлором или фтором; А1- (1S)a-циано-3-феноксибензил; (IR)a-метил-3-феноксибензил; (IR)a-этинил-3-феноксибензил; (IR) или (IRS)-циано-6-фенокси-2-пиридилметил; a-циано-3-феноксибензил IR и...
Предыдущий патент: Способ получения производных морфолина или их кислотно-аддитивных солей в виде оптических изомеров или смеси оптических изомеров
Следующий патент: Способ получения производных дигидропиридина
Случайный патент: Устройство для двусторонней поперечной обработки поверхностей расположенных друг над другом слитков