Способ получения амидинов или их фармацевтически приемлемых солей





Формула / Реферат
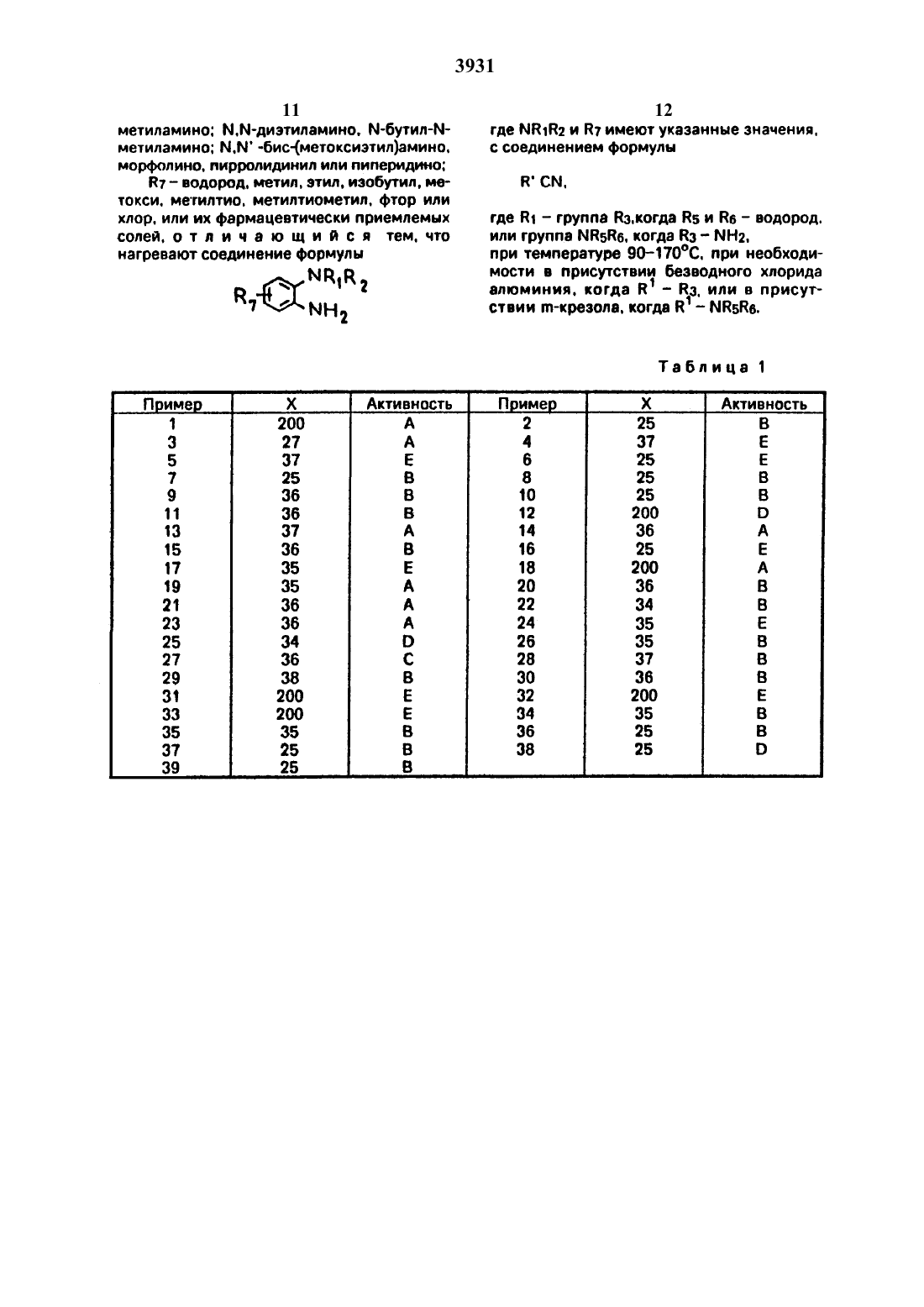
Использование: в качестве противодиабетических средств. Сущность изобретения: продукт общей формулы:
где NR1R2-морфолино, пиперидино, пирролидинил, тиоморфолино, метилпиперазинил, N,N-диметил-амино или N-(метоксиэтил-N-метиламино; R3 является алкильной группой, содержащей 1-4 углеродных атомов, когда R5 и R6 являются водородом, или R3 является группой NH2, когда NR5R6 означает N,N-диметиламино, N,N-ди-этиламино, N-бутил-N-метиламино, N,N-бис-(метоксиэтил)-амино, морфолино, пирролидинил или пиперидино; R7-атом водорода, метил, этил, изобутил, метокси, метилтио, метилтиометил, фтор или хлор. Реагент I:соединение формулы
где NR1R2 и R7 имеют указанные значения. Реагент II: R"CN, где R' является группой R3, когда R5 и R6 являются водородом, или означает группы NR5R6. когда R3 означает NH2, при 90—170°С при необходимости в присутствии безводного хлорида алюминия, когда R' означает R3, или в присутствии мкрезола, когда R'означает NR5R6. 2 табл.
Текст
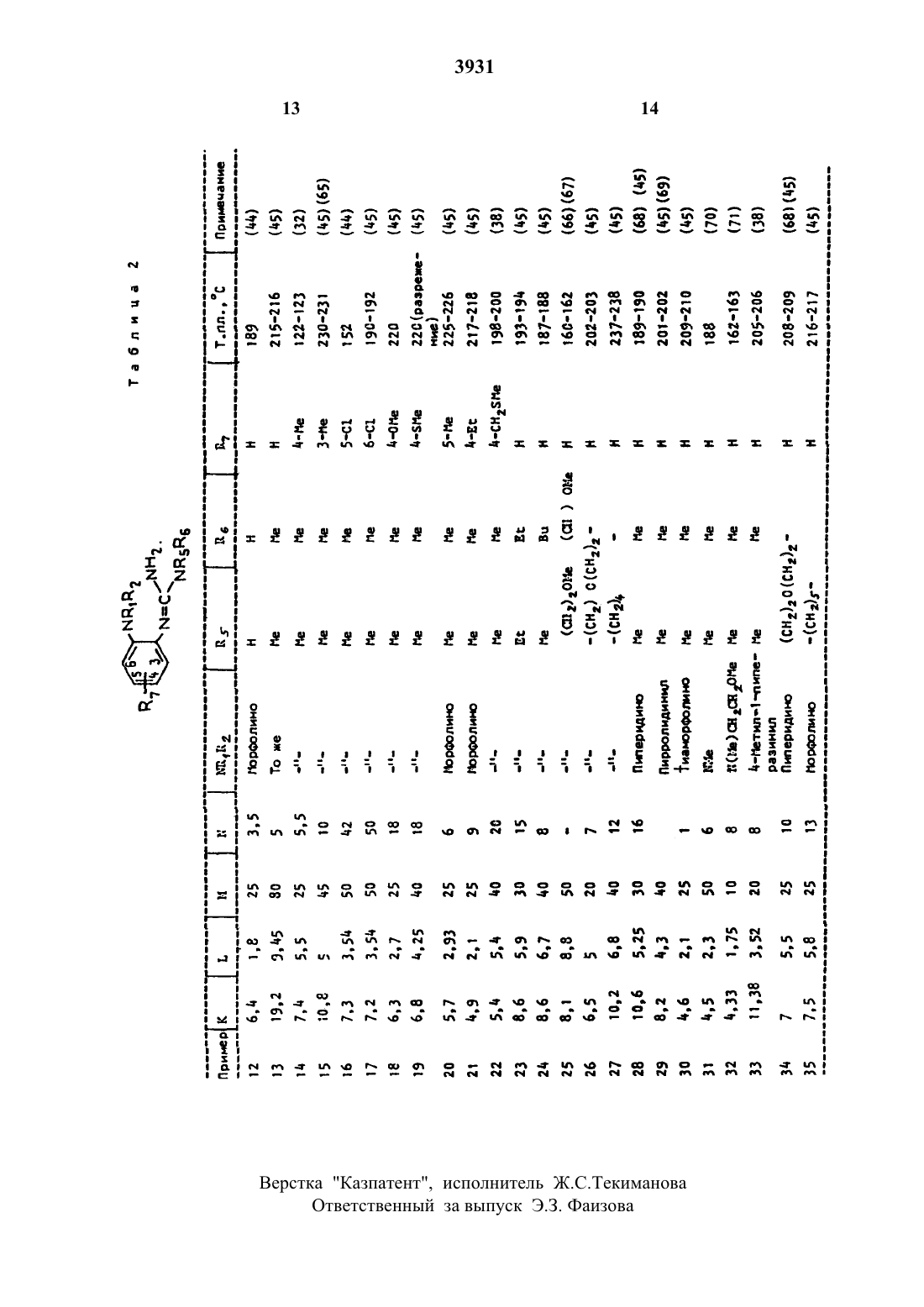
87 представляет собой атом водорода. метил. этил. изобутил. метокси. метилтио. метилтиометип, фтор или хлор.Специфическими соединениями формулы являются1.1-диметил-2-(5-метоксикарбонил-2 морфолинофенилугуанидин и их фармацевтически приемлемые СОЛИ.Соединения формулы могут существовать как соли с фармацевтически приемлемыми кислотами. Примеры таких солей включают гидрохлориды. гидробромиды. гидроиодиды. сульфаты. нитраты. малеаты. ацетаты. цитраты. фумараты. тартраты. сукцинаты. бензоаты, памоаты и соли с кислыми аминокислотами. такими как глутаминовая кислота. Соединения формулы и их соли могут существовать в форме сольватов (например. гидратов).Некоторые соединения формулы содержат один или более асимметрических углеродных атомов и существуют в различных оптических активных формах. Если соединения формулы содержат один хиральный центр. то соединения существуют в двух энантиомерных формах. изобретение включают обе энантиомерные формы и их смеси. Если соединения формулы содержат более одного хирального центра. то могут существовать в диастереоизомерных формах. Изобретение включает каждую из этих диастереоиэомерных форм и их смеси.Изобретение включает также фармацевтические композиции. содержащие терапевтически эффективное количество соединения формулы С вместе с Фармацевтически приемлемым разбавителем или носителем.При терапевтическом использовании активное соединение может быть введено перорально. ректально. парентерально или локально. предпочтительно перорально. Таким образом. терапевтические композиции согласно изобретению могут иметь форму любой из известных фармацевтических композиции для перорального. ректального, парентерапьного или локального введения. Фармацевтически приемлемые носители,пригодные для использования в таких композициях. хорошо известны в данной области. Композиции согласно изобретению могут содержать 01-90 мас. активного соединения. Композиции согласно изобретению Обычно получают в форме дозированных единиц.В качестве композиций для перорального введения используют предпочтительные композиции согласно изобретению в виде известных фармацевтических форм для такого введения. например таблетки, капсулы сиропы и водные или масляные суспензии.Средой для лекарства. используемой для получения указанных композиций. являются хорошо известные в фармакологии индифферентные составные части лекарства.Таблетки могут быть получены путем смешения активного соединения с инертным разбавителем. таким как фосфат кальция. в присутствии дезинтегрирующих агентов. например кукурузного крахмала. и смазывающих средств. например стеарата магния. и таблетирования смеси известными методами. Таблетки могут быть сформованы способом. известным специалистам в данной области. чтобы получить длительное освобождение соединений согласно изобретению. Такие таблетки. если необходимо. могут иметь знтеросолюбильное покрытие. нанесенное известными способами. например с использованием фталата ацетата целлюлозы. Аналогично капсулы. например жесткие или мягкие желатиновые капсулы. содержащие активное соединение с добавленными наполнителями или без таковых. могут быть получены традиционными средствами. могут быть снабжены растворяющимся в кишечнике покрытием известными методами. Таблетки и капсулы могут традиционно содержать каждая по 50-500 мг активного соединения.Другие композиции для перорального введения включают. например. водные растворы, содержащие активное соединение. водные суспензии. содержащие активное соединение в водной среде в присутствии нетоксичного суспендирующего средства. такого как натрийкарбоксиметилцеллюлоза. и масляные суспензии. содержащие соединение согласно изобретению в подходящем растительном масле. например в арахисовом масле.Некоторые композиции могут быть пригодны для использования соединений согласно изобретению в форме частиц очень малого размера. например. получаемых жидким помолом.В композициях согласно изобретению активное соединение может сочетаться с другими совместимыми фармакологически активными ингредиентами.Фармацевтические композиции, содержащие терапевтически эффективное количество соединения Формулы . могут быть использованы для лечения гипергликемии человека. При таком лечении количество соединении формулы 1. вводимое в день. находится в пределах 50-3000 мг. Предпочтительным путем введения является введение через рот.Г ипогпикемическую активность соединений формулы . которые представлены вфиксировали на 18 ч. затем подкожно инъецировали глюкозу (800 мг/4 мл/кг) с после ДУЮЩИМ пероральным введением ДОЗЫ соединения. подлежащего испытаниям (х мг в 4 или 5 мл 2-ного агара/кг). Через и 2 и 4 ч отбирали кровь из глазницы и глюкозу плазмы оценивали на анализаторе глюкозы Бекмана с использованием специфического метода окисления глюкозы. Затем рассчитывали процентное снижение содержания глюкозы в плазме в сравнении с контрольННМ ЖИВОТНЫМИ. КОТОрЫС НЕ ПОЛУЧЗПИ ИСПЫ тываемого соединения. но получали только 2 Ъ-ный гомогенат агара. Соединения считались обладающими гипогликемической активностью в этом тесте. если она показывала 15-ное или более снижение ГЛЮКОЗЫ ПЛЭЗМЫ при ЛЮбНХ значениях Х ДО 200 как на втором. так и на четвертом часах.По результатам, полученным при любом значении Х В ОПИСЗННЫХ выше ТВСТЗХ. оценивали гипогликемичекую активность каждого соединения, классифицировали по следующей шкале. Если для конкретного значения х имелось более одной серии результатов. то использовали среднее значение Х, снижения для классификации активности соединений.В - более чем 25-ное снижение через 2 ч. но меньше чем 25-ное снижение через 4 ч.С - снижение в пределах 15-251, через 2 ч. но более чем 25-ное снижение через 4Е - снижение в пределах 15-2536 через 2 ч. но меньше чем 15 ное снижение через 4 ч.Р - менее чем 15 Х-ное снижение через 2 ч. но более чем 15 ное снижение через 4 ч.Изобретение иллюстрируется следующими примерами. Конечный продукт каждого примера будет характеризоваться злементным анализом.П р и м е р 1. Смесь 4-(2-аминофенил) морфолина (534 г). ацетонитрила (452 мл) и безводного хлористого алюминия (12 г) нагревали при 1 б 0-170 С в течение 4 ч и получали М-(2-морфолинофенил)-ацетамидин(т.пл. 140-141 С). который перекристаллиЗОВНБВЛИ ИЗ ГВКСЗНЗ.(35 г) и безводного хлорида алюминия (12 г) нагревали при 16 О 170 С 5 ч и получали М-(5 метил-2 морфолинофениЩ-ацетамидин (т.пп. 12 С). который перекристаллизовыВЭПИ ИЗ ГВКСЗНЭ.П р и м е р 3. Смесь 4(2-аминоФенил)морфолина (д.34 г). пропионитрила (47 г) и безводного хлористого алюминия (12 г) нагревали при 160-17 ОС 6 ч и получали АНморфолинофениЩ-пропионамидин (т.пп. 114 С) который перекристаплизовывали из гексана.П р и м е р 4. Смесь гидрохлорида 4-(2 аминофенижморфолина (7.5 г) и н-бутиро нитрила (20 мл) нагревали при 170 С в герметичном сосуде высокого давления из нержавеющей стали в течение 60 ч. Избыток н-бутиронитрила удаляли, остаток растворяли в воде. подщелачивали 10-ным раствором гидроокиси натрия до рН 12 и экстрагировали дихлорметаном. Экстракт промывали водой затем рассолом, сушили и растворитель удаляли. Остаток очищали хроматографией на колонке с нейтральным глиноземом. Элюированием смесью диклорметан-гексан в отношении 11 удаляли непрореагировавший исходный материал и затем эльоированием смесью метанол-дихлорметан в отношении 199 получали твердое вещество. которое растворяли в метанопепомщиобрабатывапи фумаровой кислотой (ОД г) и получали М-(2-морфолиноФенилубутирамидин, монофумарат (т.пп. 16817 ОС). который перекристаллизовывапи из смеси метанол- эфир в отношении 12.П р и м е р 5. Измельченный в порошок безводный хлористый алюминии (12 г) по частям добавляли к перемешиваемому шламу 4-(2-ацинофенилуморфолина (534 г) и нбутиронитрила (6 г) при 5040 С. Затем смесь нагревали при 1 б 0-170 С 6 ч охлаждапи и затем вываривали с 4 О-ным водным раствором гидроокиси натрия. Раствор экстрагировали эфиром, экстракт промывали водой и рассолом и сушили. После удаления растворителя получали остаток. который кристаллизовали из смеси этилацетат гексан в отношении 11 и получали (ЧП-морфолиноФениМ-бутирамидин (т.пп. 1 З 1 С). который вращали в его монофумаровую соль (т.пп. 173 С) перекристаллизованную из пропан-2-ола.П р и м е р Б. Смесь дй-аминофенил морфолина гидрохлорида (10 г) и иэобутиронитрила (60 мл) нагревали при 1 б 5 С в течение 26 ч в герметичном сосуде высокого давления из нержавеющей стали и получали М-(2-морфолинофенил)-изобутирамидин(т.пп. 140-141 С). который перекристаллизовывали из гексане.П р и м е р 7. Смесь д-(Ъаминофенип) морфолина (534 г). изобутиронитрила (6 г) и безводного хлорида алюминия (12 г) нагревали при 1 бО-170 С 6 ч и получали М-(2-морФолиноФениЩ-изобутирамидин (т.пп. 138 С), который перекристаллизовывали из смеси этилацетат тексан в отношении 11.(32 г) нагревали при 140 С 2 ч и получали М-(Б-метилтио 2-морфолинофенил)-изобутиРЗМИДИН (т.пп. 155 С). который перекристаллизовывали из гексане.П р и м е р 9. Смесь Б-фтор-Ъморфолиноанилина (1.96 г. изобутиронитрила (2 г) и безводного хлористого алюминия нагревали при 150 С 4 ч и получали М-(5-фтор-2-морфолинофениЩ-изобутирамидин (т.пп. 142 С). который был перекристаллиэован из гексане и обращен в его фумаратную соль(т.пп. 172 С). которая была перекристаллизована из смеси метанол-эфир в отношении 11.П р и м е р 10. Смесь гидрохлорида д-(йг-аминофенипуморфолина (65 г) и валеронитрила (35 мл) нагревали при 1 б 0-1 б 5 С в атмосфере азота в течение 25 ч и затем охлаждали. Смесь обрабатывали водной гидроокисью натрия и подщепочную смесь экстрагировали дихлорметаном. Растворитель удаляли выпариванием и остаток отгоняли при давлении 50 мм рт.ст для удаления половины непрореагировавшего валеро нитрила. Твердое вещество. выделившееся при охлаждении. отделяли фильтрацией. промывали гексаном (50 мл) и перекристаллизовывали из гексана и получали М 2-мор Фолинофению-валерамидин (тпл. 1 З 5-136 С).П р и м е р 11. Смесь 4-(2-аминофенил)морФолина (356 г). пивалонитрила (5 г) и безводного хлорида алюминия (8 г) нагревали при 1 б 017 ОС в течение 6 ч и получали М-(2 морфолинофенил)-пиваламидин (т.пп. 12 бС) который был перекристаллизован из гексана и обращен в его монофумаратную соль (т.пп. 211 С). перехристаллизованную из метанола.в форме его хлористоводородной соли (К граммов) с соединением Формулы МС 3931назван. граммов) в м-крезоле (М мл) проводили нагреванием при 90-95 С в течение М часов с получением соединений. идентифицированных в табл. 1.2.П р и м е р 36. Смесь гидрохлорида 4(2-аминофенил)-морФолина (2.1 г) и ММдиметилцианамида (7 мл) нагревали в атмосфере азота при 165170 С в течение 12 ч. Реакционную смесь охлаждали до 10 С. выпавший осадок собирали фильтрацией. промывали эфиром и перемешивали с 40 ньгм водным раствором гидроокиси натрия. Полученную смесь экстрагировали дихлорметаном. экстракт промывали рассолом и сушили. После удаления растворителя получали остаток. который перекристаллизовывали из гексана и получали 1.2-диметчш 2-(2 морфолинофенил)гуанидин (т.пл. 144-145 с).П р и м е р 37. Смесь 4-(2-амино-2 метоксикарбонилфениЩ-морфолина (2.7 г). ЪДМ-диметилцианамида (1 г) и м-крезола (15 мл) нагревали при 9095 С в течение 10 ч. Добавляли лед и реакционную смесь подкисляли добавлением 2 н. хлористоводородной кислоты и полученную смесь экстрагировали эфиром. Водный слой охлаждали. подщелачивали до рН 8 добавлением твердого бикарбоната натрия и затем экстрагировали дихпорметаном. Экстракт сушили. растворитель удаляли и получали масляный остаток. который очищали хроматографией на колонке нейтрального глинозема, элюированной смесью метанол-дихлорметан в отношении 199 и получали 1,1 диметил-2(5-метоксикарбонил--морфолиноФениЩ-гуанидин (т.пл. 152154 с).(15 мл) нагревали при 9095 С б ч и получали остаток. который экстрагировали горячим гексаном. обесцвечивали древесным углем и очищали хроматографией на колонке глинозема. элюируемой смесью метанол - дихлорметан в отношении 298. Полученный продукт кристаллизовали из смеси этилацетат гексан в отношении 13. Начальный осадок удаляли фильтрацией и фильтрат выпаривали из смеси этилацетат гексан в отношении 13 и получали 12-диметип-2-(5 изобутилд-морфолинофенилугуанидин.(80 мл) и диметилцианамида (945 г) нагревали при 1 ООС 5 ч. охлаждали и добавляли к смеси 40-ного водного раствора гидроокиси натрия (300 мл) и льда (300 г). Добавляли воду (300 мл) и полученное твердоевещество выделяли фильтрацией. промывали водой и растворяли в дихлорметане. Раствор сушили. растворитель удаляли и ПОЛУЧЗПИ ОСТЗТОК. КОТООЫЙ ПЭОЗКОИГЭЛПИ зовывали из гексана и получали (д-диметил-2-(2 чиорфолинофенит-гуанидин (тлл. 142143 с).П р и м е р 40. К размещенному раствору М-этил-М-О-метоксиэтил)-амина (1 О.3 г) в безводном бензоле (80 мл) охлажденном до 5 С в ледяной бане. в течение 10 мин добавляли раствор бромистого циана (533 г) в безводном бензоле (20 мл) и температуру реакционной смеси доводили до комнатной После этого реакционную смесь размешивали в течение ночи. Затем растворитель удаляли при пониженном давлении. а остаток неоднократно экстрагировали безводным эфиром (4 х 25 мл). Органические экстракты осушали сульфатом натрия. фильтровали, растворитель удаляли. в результате получали М-этил-М-(Ъметоксиэтил)цианамид в виде бледнсгжелтого маслянистого продукта 6.3 г).Смесь М-(2-аминофениЩ-морфолина гидрохлорида (2 г). М-этил-М-Ш-метоксиэтилндианамида (2.6 г) и м-крезола (15 мл) нагревали при 9 КЪ 95 С в течение 8 ч. Затем добавляли М-этил-М 2-метоксиэтил)циана мид (2.5 г) и продолжали нагревать еще в течение 20 ч. После удаления растворителя получали желтоватое маслянистое вещество. которое очищали с помощью колоночной хроматографии на нейтральной окиси алюминия (80 г). эпюируя сначала дихлорметаном, а затем 1-ным метаноломдихлорметаном, в результате чего получали бесцветное маслянистое вещество (1 г). которое затем подвергали реакции взаимодействия с фумаровой кислотой (д.38 г) в метаноле (Ю мл) и получали М-этил-Н-(Ъметоксиэтил)-М-24-морфолино)-фенил-гуанидина монофумарат в виде бесцветного твердого вещества (0.В г. т.пл. 17 О-171 С). которое перекристаллизовывали из смеси метанола и эфира (122), Формула изобретенияСпособ получения амидинов общейПз- алкильная группа содержащая 1-4 атомов углерода. когда 85 и Не - водород. или На - группа МН 2. когда Мкздв ММ-ди
МПК / Метки
МПК: C07D 295/135, A61K 31/445, C07C 257/14
Метки: способ, получения, фармацевтически, приемлемых, амидинов, солей
Код ссылки
<a href="https://kz.patents.su/7-3931-sposob-polucheniya-amidinov-ili-ih-farmacevticheski-priemlemyh-solejj.html" rel="bookmark" title="База патентов Казахстана">Способ получения амидинов или их фармацевтически приемлемых солей</a>
Способ получения ( 2 – морфолинофенил) гуанидинов или их фармацевтически приемлемых солей

Номер патента: 2848
Опубликовано: 15.12.1995
Автор: Баласубраманьян Гопалан
МПК: A61K 31/535, C07D 295/135
Метки: приемлемых, способ, солей, фармацевтически, гуанидинов, получения, морфолинофенил
Формула / Реферат:
Использование: в качестве противодиабетических средств в медицине. Сущность изобретения: продукт (2-морфолмнофенил)-гуанидины общей формулы где NR1R2 - метиламино, диметиламино, этиламино, метилэтиламино, бутиламино, аллилметиламино, этилметоксиэтиламино, диаллиламино, N'-метилпиперазинил, диметилморфолино, метилпиперидинил, морфолино или тиаморфолино и R3-Н, F, CI. СН3 или СН3О, или их соли. Реагент 1: HNR1R2. Реагент 2:...
Способ получения сульфинильных производных гетероциклических соединений или их фармацевтически приемлемых солей

Номер патента: 1863
Опубликовано: 15.03.1995
Авторы: Девид Кокс, Джон Льюис Сучитский, Энтони Говард Инголл
МПК: C07D 235/28
Метки: способ, фармацевтически, получения, гетероциклических, соединений, приемлемых, сульфинильных, производных, солей
Формула / Реферат:
Изобретение касается гетероциклических соединений, в частности их сульфиннльных производных общей формулы M-S(0)-Rc (I), где М - группа формулы (II), а Rс - группа формулы -(СR10 R17)у - (CR18R19)z-Rx, где Y и Z (равные или разные) - 0 или 1, или 2; R16,R17 и R19-Н и Rх- группа формул (III), (IV) или (V):и, когда (Y+Z) не = 0, Rx может быть NR9R10 R2, R3, и R4 (равные или разные) - II, галоген, алкокси, алкил или -N(R)2 , или смежная пара R1,...
Способ получения правовращающих производных оксиизоиндолинила или их фармацевтически приемлемых солей

Номер патента: 1561
Опубликовано: 15.12.1994
Авторы: Фабрицио Орци, Джованни Карниель, Пьерлуиджи Гриджи, Джорджио Черони, Бруно Миорини
МПК: C07D 209/46
Метки: солей, оксиизоиндолинила, способ, получения, приемлемых, производных, фармацевтически, правовращающих
Формула / Реферат:
Изобретение касается гетероциклических веществ, в частности правовращающих производных оксииэоиндолина общей формулы I где Х = C6H4-пaрa-CHR-C(O)-O-R1 ; R=C1-С4-алкил; R=H или С1-С4-алкил, или их фармацевтически приемлемых солей, обладающих обезболивающей и противовоспалительной активностью, что может быть использовано в медицине. Цель изобретения - повышение выхода целевого продукта. Синтез ведут реакцией офталевого ангидрида с правовращающим...
Способ получения хинолинкарбоновых кислот или их фармацевтически приемлемых солей

Номер патента: 2465
Опубликовано: 15.09.1995
Авторы: Делле Вашвари, Петер Ритли, Иштван Хермец, Мария Балог, Агнеш Хорват, Геза Керестури
МПК: A61K 31/4, C07D 401/04
Метки: хинолинкарбоновых, приемлемых, способ, фармацевтически, солей, получения, кислот
Формула / Реферат:
Изобретение касается гетероциклических веществ и, в частности, получения хинолинкаобоновых кислот общей формулыгде R-пиперазинил, 4-метилпиперазинил, 4-этилпиперазинил, или их фармацевтически приемлемых солей, обладающих антибактериальной активностью и используемых в медицине. Цель изобретения - повышение выхода целевого продукта и сокращение времени процесса при лучшей его избирательности. Синтез ведут реакцией соответствующих производных...
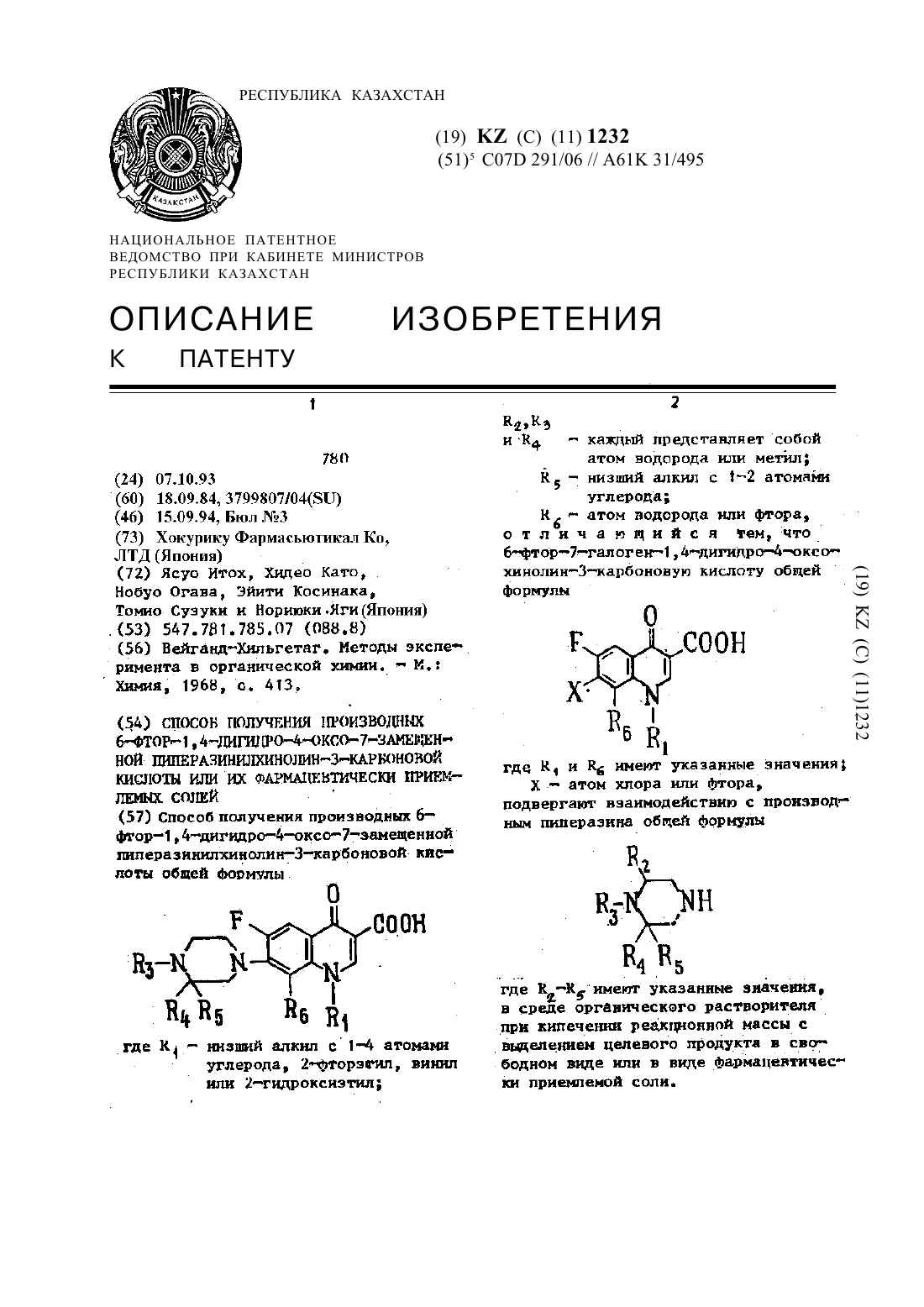
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты или их фармацевтически приемлемых солей
Номер патента: 1232
Опубликовано: 15.09.1994
Авторы: Хидео Като, Эйити Косинака, Нориюки Яги, Ясуо Итох, Нобуо Огава, Томио Сузуки
МПК: A61K 31/495, C07D 401/02
Метки: получения, производных, приемлемых, солей, 6-фтор-1,4-дигидро-4-оксо-7-замещенной, кислоты, пиперазинилхинолин-3-карбоновой, способ, фармацевтически
Формула / Реферат:
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты общей формулыгде R1 - низший алкил с 1-4 атомами углерода, 2-фторэnил, винил или 2-гидроксиэтил; R2, R3 и R4 - каждый представляет собой атом водорода или метил; R5 - низший алкил с 1-2 атомами углерода; R6 - атом водорода или фтора, отличающийся тем, что 6-фтор-7-галоген-1,4-дигидро-4-оксо-хинолин-3-карбоновую кислоту общей формулыгде R1...
Предыдущий патент: Способ обогащения сульфидных руд
Следующий патент: Устройство для перемешивания молока и воздуха
Случайный патент: Способ реконструктивной мастоидопластики после санирующих операций на ухе