Способ получения производных цефалоспорина или их легко гидролизуемых сложных эфиров, или их солей с щелочными металлами






























Формула / Реферат
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЦЕФАЛОСПОРИНА ИЛИ ИХ ЛЕГКО ГИДРОЛИЗУЕМЫХ СЛОЖНЫХ ЭФИРОВ ИЛИ ИХ СОЛЕЙ С ЩЕЛОЧНЫМИ МЕТАЛЛАМИ формулы I
в виде син-изомеров, где R1 - атом водорода, С1-С2 алкил, возможно замещенный атомом брома, карбоксиметил, этоксикарбонил, бензил, бензоил, 1-оксигексадецил или циклододецилоксикарбонил, R2 - атом водорода, остаток легко гидролизуемой сложно-эфирной группы или катион щелочного металла, такого как натрий, R3 - прямой или разветвленный С1-C4 алкил, возможно прерванный атом кислорода, аллил или бензил, X - атом кислорода или серы или сульфинил, n - 0 или 1, отличающийся тем, что соединение формулы II
где R3, Х и n имеют указанные значения, подвергают взаимодействию с реакционноспособным производным кислоты формулы III
где R1 - 1-метил-1-метоксиэтил, С1-С2, алкил, возможно замещенный атомом брома, бензил, бензоил, 1-оксигексадецил или циклододецилоксикарбонил, в среде инертного органического растворителя в присутствии основания при температуре от -70° С до комнатной и в полученной продукте удаляют защитные тритильную и 1-метил-1-метоксиэтильную группы или предварительно удаляют 1-метил-1--метоксиэтильную группу, этерифицируют гидроксильную группу с получением ее простого трет-бутоксикарбонилметилового эфира или сложного эфира этоксикарбоновой кислоты с последующим удалением тритильной группы в случае необходимости с одновременным переводом простого трет-бут-оксикарбонилметилового эфира в простой карбоксиметиловый эфир и выделяют целевой продукт в виде свободной кислоты или ее cложного легко гидролизуемого эфира или ее соли с щелочным металлом, таким как натрий.
Текст
окснкарбонилметнлового эфира в простой карбоксиметнловый эфир и вьщелнют целевой продуктв виде свободнойполучения иовьшантибиотиков Цвфалот споринового ряда, а именно новых проНЗВОДНЬК ЦфаЛОСПОрНН-а РЕП ИХ ЛЕГКО гндролизуемсложныш эфиров или их СОЛЕЙ С ЩЕЛОЧНЬЧ МЕТЗЛГЛЗНЭ, КОТОРЫЕ обладают антимикробной активностью и могут найти применение-в медицине.-Цель изобретения т получение нот вьш антибиотиков цефалоспоринового ряда, обладающих повышенной-активностью как против грамположительных,так н против грамотрицательньш бант терии в тон числе устойчивы по отНОШЕНИЮ Н НЗВЕСТНЪШ ННТНЬНОТНКЗМ- П р и м е р 1 3-Метоксиметнл 72(2 амнотиазолтд-ил)-2-(гидрок сиимино)ацетамидотцеф 3 емч 4 карбоСуспенцнруют 3,01 г солн триэтнл амина 2 д(2 тритиламинотиазолнп 3-2(1-нетил 1 метокснэтоксиимно) уксусной кислотьь синизонера в 15 си ацетона. Прибавляют 1,05 г тозилхлорида и перемешивают в течение 1,5 ч. Вводят-20 см этилового эфира вриесь, охлаждают ДО н 10 С,отсасывают, промывают эфиром и получают 2,90 г продукта, состоящего из целевого ангидридаи хлортидратаРастворяют 0,732 г 4 аммно 3 нет окснметипцеф 3-ем-4-карбоновой кисло ты в 10 си хлористого метилена н 0,84 см триэтиланнна, охлаждают до-20 С и прибавляют 2,4 г полученной в стадии А смеси, дают температуре подняться до комнатной, прибавляют 0,5 см уксусной кислоты промваюткислоты или ее сло. эго легко гидролизуемого эфира или ее соли р щелочным металлом, таким как натрий.-водои, сушат, концентрируют досуха, растирают с этиловьм эфиром, отсасьг вают и получают 3,07 г-сырого продукта, который перекрнсталлизуют В метаноле для получения 1,21 г цепед вого продукта. дПри 4550 С перенешвают в тече-. иие 10 мин 1,1 г полученного продукта с 5 см водной муравьиной кислоты прибавляют 2 си 3.воды, отсасывае ют, концентрируют досуха фильтрат под уменьшенным давлением при 307 С,отгоняя воду этанолом. Остаток крнсталлизуют в воде. Отсасывают н полУчают 0,54 гсырого продукта, ко торыи растворяют при помощи тризтнлд амина в 5 см 502 ного водного эта нола. Подкисляют до рН 2-3 прибавлением муравьиной кислоты, отсасывают кристаллнэоваиный продукт, цромвают этанолом, а затеи эфиром и получают 0,45 г сольвата. .Стадия А 1 оксопропокснметнлоВый ЭФИР 3-метоксиметнл 7-2(2-трнтипаминотнаэол-4-ил)-2 т(т-метнл-1-метоксн)-этоксиимнно/ацетамн доЦеф-3-ем-4-карбоновой кислоты,синизонер. и При комнатной температуре растВОРЯЮТ 4,15 г 3-нетокснметил 7-2(2-трнтиламмнотназол-4-ил-2-(1-метокснметнл/-этоксннмно)-ацетаммдо-цеф-3-ем-4-карбоновой кислоты, снн-изомер, и 0,456 г сухого карбоната калия в 14 см 3 безводиого днметилформамица. 0 хлаждают.до 0 С,вводят в течение 10 мин приготовленную, как это указано нне, суспен зию иодметилового эфира пропионовойкислоты н перемеивают 30 мин при 0 С, а затем 30 мин при 20 С. Реакционную среду выливают в смесь, состоящую из 340 си воды, 17 см нор-мального водного раствора бикарбоната натрия и 50 см этилового эфира уксусной кислоты. Переешивают декантируют, экстрагируют этиловым эфиром уксусной кислоты, промвают водой, сушат, концентрируют досуха под уменьшенным давленем при температуре иже 35 С. Остаток забирают хв 25 см изопропилового эфира и отсасывают 4,42 г целевого продукта.ямт (СВС 1,), ч./или 1,15 (К, 1 7), 2,40 (4, 18 ,-С 2 Н 5, 3,34 сосну 3.5515 зонд) 4,33 (сн,осн,) 5,05 (а, 1 н 5 Н 6) 6,71 (Н, син-тиазола) 7,33 (тритии). Получение иоднетилового эфира пропионовой кислоты.нагревают с обратным колодилвнн- Жом втенение 10 ни 1,4 г кларнетилового эфира пропионовой кислоты,2,71 г йодида натрия н 23 см безводного ацетона. Получают суспензию,которую употребляют сразу.раствора нуравьинойкислоты. Разбавпяют с 90 см воды в горячем состоянии, отсасывают и отгоняют фильтрат под уменьшенным давлением при температуре ниже 30 С. Остаток эабирают в 100 см хлористого нетнпена промываютразбавленньм до 1/10 насыщенным раствором хлористого Натрия н7 смз нормального раствора бикарбоната.иатрил а затем разбавленным до 1/10 насьшенным раствором хлористого натрия. Органический слой сушат, перегоняют досуиа под уменьшенным давлением н остаток забирают в 1 О 0 см этилового эфира, отсасывают н полу чают 2,10 г сырого продукта. Забирают его в 15 см этилового эфира уксусной кислоты, перемешивают30 мин, отсасывают,-прополаскивают этиловым эфиром уксусной кислоты, а затем этиловым эфиром и получают 1,69 г продукта. Растворяют 1,57 г этого продукта а 15 см хлористого метилена, отфнльтровышают, перегоннют под уменьшенным данлениен,остаток забирают в 10 см этилового эфира уксусной кислоты, леремешнают30 мни, отсасывают, прополаскивают этиловым эфиром уксусной кислоты, а затем эфиром и получают 1,29 г целевого продукта -перемешивают 1,80 г трнэтиланновой соли 2-(2-тритиламмиотиазол-6-пл)-2-нетоксиииноуксусной кислоты, сии-изомер, и 0,63 г хлорида тоэнла в 20 см безводного ацетона. Перемешивают в течение 1 ч при 20 С и получают суспенэию,.которал сразу употребляется на следующей стади.чприготавливают перед унотребле ниен следуюцм раствор при 2 ОС при5. перемешиванн в инертной атмосфере 0,732 г 3-метоксиметнл-7-амино-цеф-З-ем-А-карбоновой кислоты, 6,6 см молярного раствора бикарбопата натрия и 3,4 см водим Охлаждают до 5 с.и за 5 мин вводят полученную встадим А суспензию смешанного ангидрида. Перемешивают1 ч при 0-5 С а затеи 1 ч при 20 с. Отфнльтровывают нерастворимое вещество и перегоняют ацетон под уменьшеинындавлениеи при Максимум 30 С. Подкнсляют прибавиой 0,7 см муравьиной кислоты, экстрагируют хлорис тым метиловом, промывают водой, су- шат иконцентрнрукп-досуха под уменьшенным давленеи. Остаток заби рают В 10-си этилового эфира, отсасывают и получают 1,75 г продукта,Кбторнй Употребляют в данном виде на чледущцей стадии. - .кислота, сип-изомер, ПРИ 45-50 С в инертной атмосфере Перемешивают втечение 12 мн 107 г П 0 ПУченного.на предыдущей стадии продукта и 8,4 сн 661 ной водной муравьиной кислоты, при 45-50 С прибавляют 3,4 см-воды и сразу отсасывают. Фнльтррт перегоняют под уменьшенным давлением, сушат перегонкой с зтанслон, растирают сухой,экстракт с 10 см воды. отсасывают,прополаскивают водой, а затем этиловым эфиром и получают 0,436 г целевопо продукта. из неточным растворовМедленноприбавляют 0,83 си молярно о раствора бикарбоната натрия, раз бавляют 1 см 2 М раствора хлористого натрия, отсасывают, прополаскивают водой Фильтрат подкисляют0,5 сн 2 н. раствора соляной кислоты до рН 3,отсасывают, прополас кивают водой, эфиром, сгущают ацетонони получают 0,227 г очищенного продукта. -и 2,8 см триэтиламниа,-охлаждают до 20 С н вводят 9,5 г паратолуопсуль фонового 2-(2-1 ритипаминотиаэал-4-ил)-2 ч(1-петил-1-метоксиэтоксимно) уксусного ангидрида, полученное гокак это указано в стадии А примера 1. Перемешвают при 0 С в течение 2 ч, подкисляют прибавкой 1,5 см уксусной кислоты, промывают водой органический слой, сушат и концентрируют досуха под уменьшенным давлением. Остаток забирают в 50 см этиловогозфира, отсасывают,прополаскивают эфиром н получают 8,65 г сырого продукта. Забирают его в 45 см метанола и перемешивают в течение 30 мин. Затравляют кристаллизацию, отсасывают, прополаскивают нетанолон, а затем этиловым эфиром и получают 5,2 г-целевого продукта. -При 50 С и в течение 10 мин перемешшвают 3,72 г полученного выше про,дукта и 18,6 см 662 ной водной муравьиной кислоты. Прибавляют 7,4 см воды и.сразу отсасывают. Фильтрат перегоняют под уменьшенным давленн ем при максимум 30 С, перегоняют- 12 раза со смесью этанол-вода (2-1), забирают остаток в 10 смд воды,от сасывают, прополаскивают водой, а затем этиловьм эфиром и получают 19 д 5 г сырого продукта. Забирают его в 136 см 502-кого водного эта нона н медленно прибавляют 0,63 см тризтиламщна. Отсасывают нерастворимое вещество, подкнсляют фильтрата эатем этиловн эфиром и получают 1,392 г очищенного продукта.иминоацетамидоцеф-3-емтд-карбоно вой кислоты, сии-изомер. . Стадия А оксопропоксиметвловыи эфир 3-метнотиометил 7 Е 2 т(2 тритнламнотиаэол-4-ил)-2-(1-метил-1-метоксиэтоксннмино)ацетамдо-цеф 3-ем 4 кврбоновой кислоты, син-изомер. При 20 С перемешивают 5,2 г 3-метилтиометил-7-2-(2 тритнламмнотиазол 4 ил)2(1-метнлтнетоксн-этоксниммно)ацетамидоцеф-3 ем 4 ч карбоновой кислоты, синизомер, и 0,56 г карбоната калияв 20 см безводного диметилформанде, охлаждают до 5 С иррнбавпят при 5-тос 28 смд ацетоновой суспензии йодометнлового эфира пропионовой кислоты, 8приготовленной перед употреблением,исходя из 1,75 г клорметилового эфира пропионовой кислоты. Перемешивают 30 мин при 5 С, а затем 30 мин при 20 СРеакционную смесь выливают в раствор, обраэованнът 260 см воды при 1015 С, 20 см н. водного раствора бикарбоната натрия и 50 см этилового эфира уксусной кислоты, перемешивают, декантируют, экстрагируют этиловым эфиром уксусной кислоты,промывают водой, сушат и перегоняют досуха под уменьшенным давлением. Остаток забирают в 50 см изопропилоПри 50 С в течение 10 мин перепет шивают 3,75 г полученного въше продукта в 18,7 см 662 ной водной муравьиной кислоты. Прибавляют 7,5 см воды, отсасывают, перегоняют фильт РЕТ ПОД УМЕНЬШВННЫМ давлением примаксимум 30 С н производят две пере гонки со смесью этанол-вода (2 т 1). Остаток забирают в 10 см воды, отсасывают, прополаскивают водой, а затем этиловым эфиром и получают 2,17 г сырого продукта.-Этот последний хроматографируют на двуокиси кремнии, элюируют смесью этиловый эфир уксусной кислоты ацетон (3-1),эабирают сухой остаток в 10 см изопропилового эфира, отсасывают4,5 сн этилового эфира уксусной кислоты,-прибавляют 7,5 см этилового эфира уксусной кислоты, отсасывают, прополаскивают этиловым эфиром уксусной кислоты, а затем изопропи ловым эфиром н получают 0,536 г це левого продукта.
МПК / Метки
МПК: A61K 31/545, C07D 501/06
Метки: солей, способ, сложных, гидролизуемых, получения, эфиров, цефалоспорина, щелочными, легко, металлами, производных
Код ссылки
<a href="https://kz.patents.su/30-1574-sposob-polucheniya-proizvodnyh-cefalosporina-ili-ih-legko-gidrolizuemyh-slozhnyh-efirov-ili-ih-solejj-s-shhelochnymi-metallami.html" rel="bookmark" title="База патентов Казахстана">Способ получения производных цефалоспорина или их легко гидролизуемых сложных эфиров, или их солей с щелочными металлами</a>
Способ получения производных 13-галоидмилбемицина или их солей, или их сложных эфиров

Номер патента: 1235
Опубликовано: 15.09.1994
Авторы: Акира Нисида, Тосиаки Янаи, Энтони О" Салливан, Бруно Фрай, Кацуо Сато, Норитоси Китано
МПК: A01N 13/02, C07D 493/22
Метки: производных, эфиров, сложных, солей, способ, получения, 13-галоидмилбемицина
Формула / Реферат:
Изобретение относится к гетероциклическим соединениям, в частности к получению производных 13-галоидмилбемицина фор-лы I лы =N-OR2, где R2-Н, С1-С6 - алкил, или их солей, или их сложных эфиров, которые проявляют антигельминтную, акарицидную и инсектицидную активность.Цель - разработка способа получения новых более активных соединений. Получение их ведут галогенированием соединения фор-лы I, где R1 указано выше, в положении 5- O или ОН и Н, а в...
Способ получения эфиров – производных циклопропанкарбоновой кислоты

Номер патента: 1552
Опубликовано: 15.12.1994
Авторы: Андре Теш, Жан Тессье, Жак Мартель
МПК: A01N 53/00, C07C 69/743
Метки: производных, циклопропанкарбоновой, кислоты, способ, эфиров, получения
Формула / Реферат:
Изобретение касается производных циклоалифатическнх кислот, в частности получения эфиров замещенной циклолропанкарбоновой кислоты общей формулы R-O-C(O)-CH=CH-СН-С(СН3)2-СН-С(О)OА1 , где R-C1-C4-алкил, замещенный хлором или фтором; А1- (1S)a-циано-3-феноксибензил; (IR)a-метил-3-феноксибензил; (IR)a-этинил-3-феноксибензил; (IR) или (IRS)-циано-6-фенокси-2-пиридилметил; a-циано-3-феноксибензил IR и...
Способ получения производных 2-тиенилоксиуксусной кислоты или их фармацевтически приемлемых солей

Номер патента: 1567
Опубликовано: 15.12.1994
Авторы: Дитер Биндер, Хуберт Петер Фербер, Франц Ровенсцки
МПК: C07D 333/26, A61K 31/38
Метки: фармацевтически, 2-тиенилоксиуксусной, солей, способ, приемлемых, получения, кислоты, производных
Формула / Реферат:
Изобретение касается производных 2-тиенилоксиуксусной кислоты, в частности получения 5-[2-(бензолсульфониламино)этил]- или 5-[2-(4-хлорбензолсульфониламино) этил]-2-тиенилоксиуксусных кислот, которые могут быть использованы в медицине для лечения тромботических заболевании. Цель - создание новых более активных веществ указанного класса. Синтез ведут окислением, например, амида N-[2-[2-(5-(2-гидрокси)-этокси)-тиенил]-этил]-бензолсульфоновой...
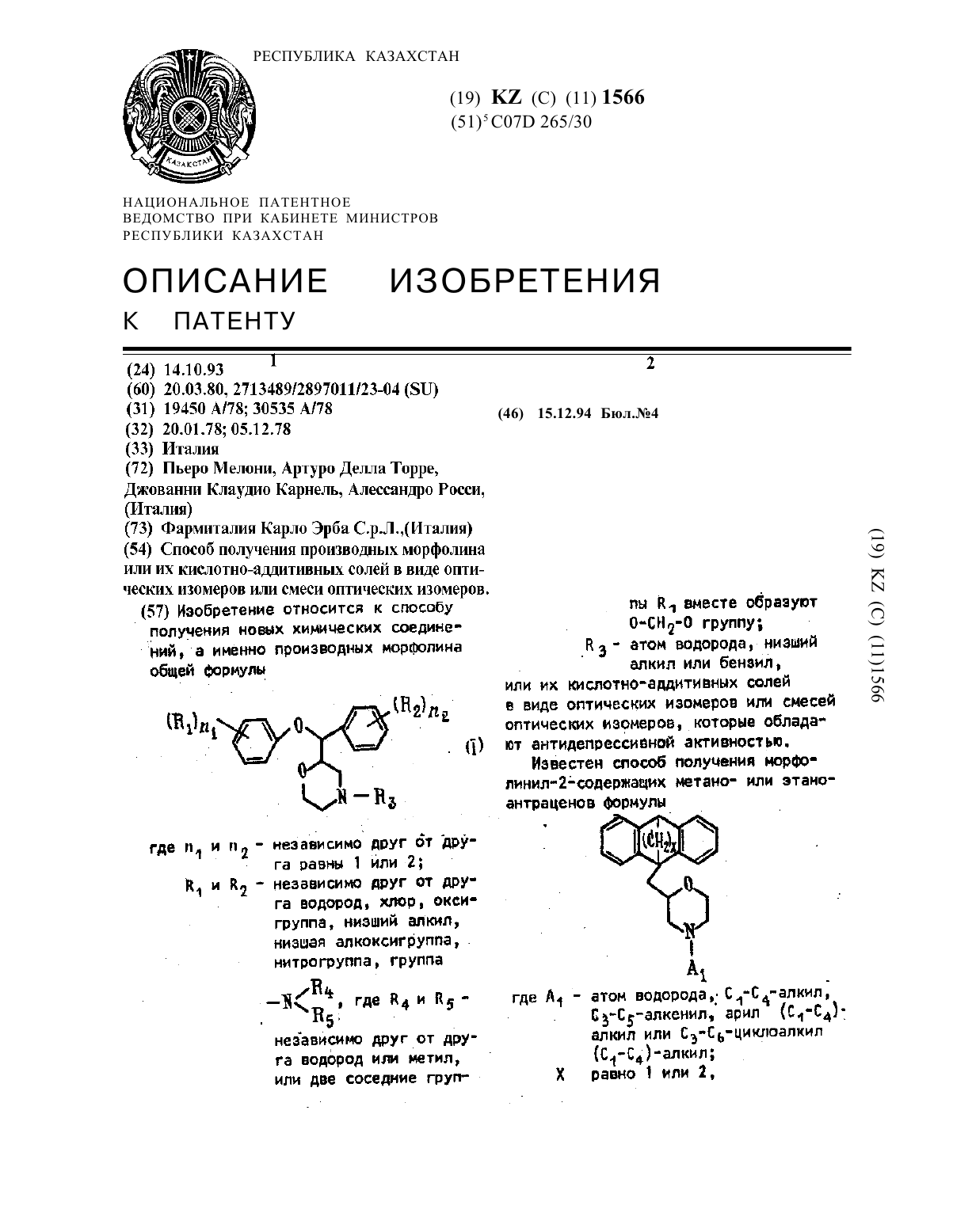
Способ получения производных морфолина или их кислотно-аддитивных солей в виде оптических изомеров или смеси оптических изомеров
Номер патента: 1566
Опубликовано: 15.12.1994
Авторы: Пьеро Меллони, Алессандро Росси, Артуро Делла Торре, Джованни Клаудио Карнель
МПК: C07D 265/30, A61K 31/535
Метки: морфолина, изомеров, производных, кислотно-аддитивных, солей, оптических, получения, способ, смеси, виде
Формула / Реферат:
Изобретение относится к способу получения новых химических соединений, а именно производных морфолина общей формулы где n1 и n2- независимо друг от друга равны 1 или 2; R1 и R2 - независимо друг от друга водород, хлор, оксигруппа, низший алкил, низшая алкоксигруппа, нитрогруппа, группа где R4 и R5 - независимо друг от друга водород или метил, или две соседние группы R1 вместе образуют О-СН2-О группу, R3 - атом водорода, низший алкил или бензил,...
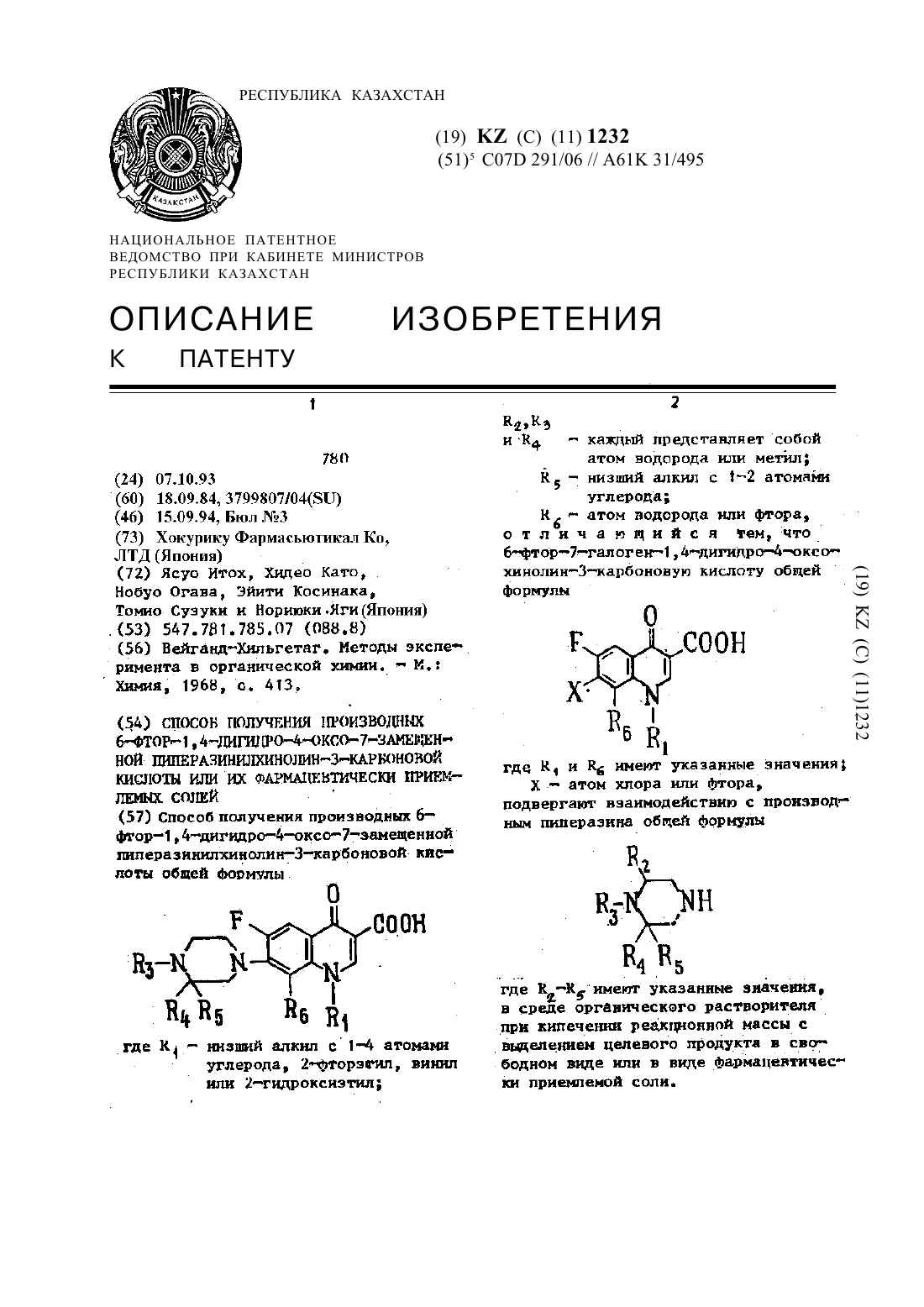
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты или их фармацевтически приемлемых солей
Номер патента: 1232
Опубликовано: 15.09.1994
Авторы: Нориюки Яги, Ясуо Итох, Хидео Като, Эйити Косинака, Нобуо Огава, Томио Сузуки
МПК: A61K 31/495, C07D 401/02
Метки: приемлемых, пиперазинилхинолин-3-карбоновой, способ, кислоты, солей, фармацевтически, производных, получения, 6-фтор-1,4-дигидро-4-оксо-7-замещенной
Формула / Реферат:
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты общей формулыгде R1 - низший алкил с 1-4 атомами углерода, 2-фторэnил, винил или 2-гидроксиэтил; R2, R3 и R4 - каждый представляет собой атом водорода или метил; R5 - низший алкил с 1-2 атомами углерода; R6 - атом водорода или фтора, отличающийся тем, что 6-фтор-7-галоген-1,4-дигидро-4-оксо-хинолин-3-карбоновую кислоту общей формулыгде R1...