Способ получения сульфинильных производных гетероциклических соединений или их фармацевтически приемлемых солей
Номер патента: 1863
Опубликовано: 15.03.1995
Авторы: Девид Кокс, Энтони Говард Инголл, Джон Льюис Сучитский









Формула / Реферат
Изобретение касается гетероциклических соединений, в частности их сульфиннльных производных общей формулы M-S(0)-Rc (I), где М - группа формулы (II), а Rс - группа формулы -(СR10 R17)у - (CR18R19)z-Rx, где Y и Z (равные или разные) - 0 или 1, или 2; R16,R17 и R19-Н и Rх- группа формул (III), (IV) или (V):
и, когда (Y+Z) не = 0, Rx может быть NR9R10 R2, R3, и R4 (равные или разные) - II, галоген, алкокси, алкил или -N(R)2 , или смежная пара R1, R2, R3 или R4 могут вместе с атомом углерода образовать ненасыщенное карбоциклическое или N-содержащее кольцо с 6-ю членами, Х - О, S или NH, R -Н или алкил; R5, R6, R7, и R8-(равные или разные) - II галоген, алкокси, алкил или-N(R)2; R9 и R10 (равные или разные) - II, алкил, циклоалкил или R9 R10, — насыщенное или ненасыщенное кольцо с 4-8-ю членами (оно может иметь 0,1 или 2 гетероатома и 1 или более заместителей R1),или R9- Н, а R8 и R10 вместе с атомами азота и углерода кольца, к которому атом азота и R1 присоединены, образуют кольцо с 4-8-ю членами (оно может иметь 0,1 или 2 гетероатома и 1 или более заместителей R1); А - N- или S-содержащее кольцо с 5-ю или 6-ю членами, которое присоединено к остатку молекулы кольцевым атомом углерода, при условии, что а) Rс не = -СН2-CH2 - морфолино, б) когда Х - NH, то Rc не содержит ненасыщенное N-содержащее кольцо, кроме кольца, замещенного замещенной или незамещенной NH2, в) когда Х - >NH, то Rc не содержит алкил, замещенный необязательно алкилом или галозамещенной пиперидино-группой, или его фармацевтически приемлемой соли, которые ингибнруют желудочную кислотную секрецию. Цель изобретения - создание новых более активных соединений указанного класса Синтез ведут избирательным окислением соответствующего соединения формулы (VI) M-S-Rc, где М и Rс- см. выше, в органическом растворителе с выделением целевого продукта в свободном виде или в виде фармацевтически приемлемой соли. Новые соединения имеют эффективную позу ED50 = 2,5-55 мМ при низкой токсичности, против ED50= 80 мМ для тимепразола.
Текст
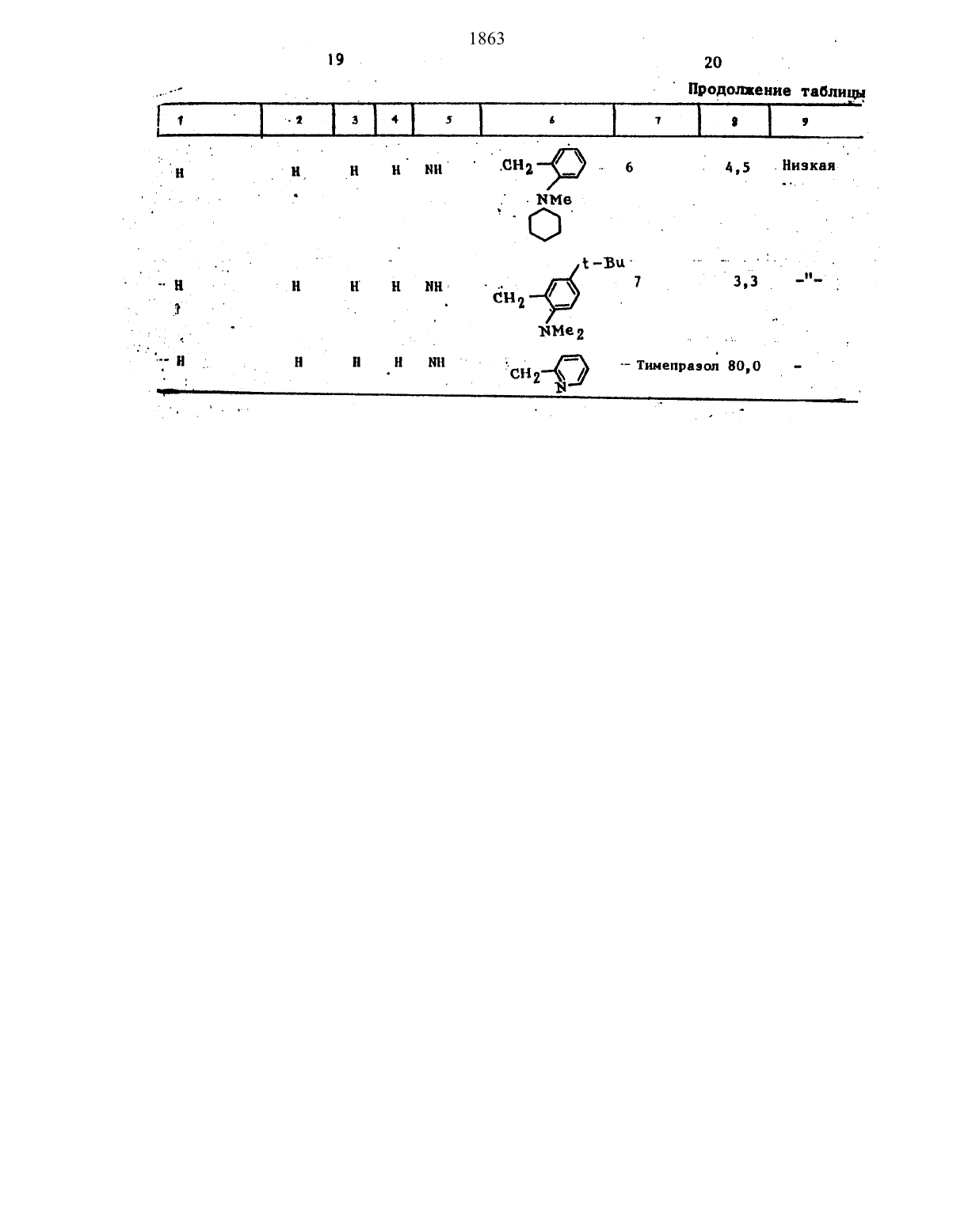
виде или в виде фармацевтически приемлемой соли. Новые соединения имеют эффективную позу ЕВ. 2,5-55 мнизобретение относится-кспособу получения новых супьфинильных производных гетероциклических соединений и к их фармацевтически приемлемьм солям,которые предупреждают или ингибируют желудочную кислотную секрецию.гцель изобретения синтез поцьщ соединений,-по своей активности превосходящих структурный аналог.Гндрохлорнд 2 диетиламннобензил-. хлорида (818 г) растворнют в сухом диметилформамиде (100 мл), обрабаты-. вают-2-меркаптобенаимидазолом (5 д г) ибеэаоднымдкарбоиатомокалняЧ(11,0 г) и полученную смесь перемешивают прн комнатной температуре в течение ночи. Реакционную смесь вливают в воду н экстрагируют этилацетатом, который промывают водойн сушат над сульфатом магния. Растворитель выпаривают и остаток.перекристаллизовывают из толуола с получением 568 г твердого.вещества кремового цвета. Продукт элюируют из испарительной хроматографи.ческой колонки смесью дихл 0 РМгТа ютилацетат (91).в качестве ЭЛЮЗНТН с получением бесцветного твердого вещества с т.пл. 158-16 ОС..982-ную м-хлорпербензойную кислоту (0,6 г) добавляют порциями в течение нескольких минут к перемешиваемому раствору продукта а (10 г) в дихлорметане (30 мл) при 0 С. Реак ционную смесь перемешивают в течение 0,5 ч, промывают насыщенньм воднымраствором бикарбоната натрия, затем соляным раствором и сушат. Растворнтель ВЪШЗРИВЗЮТ Н ОСТЗТОК ЗЛХОНРУЮТ НЗ испарительной хроматографической колонки с использованием в качестве элюанта.смесь дихлорметан-этилацетат(73)с получением 0,6 г бесцветного твердого вещества с т.пл. 120121 СвПр и м е р 2. С помощью способа,описанного в примере 1, с использованием соответствующихисходньш неществ могут быть приготовлены следующие соединения 2-(д-метокси-3,5-днметил-2-пнрндиннлметнлсульфиннп)бензоксазол (а), т.пл. 1 о 3-105 с 2-(4-метоксн-3,5-диметнл-2-пнрндинил Мтнлсулъфинил)-бенотиазоп(Ь), т.пл. 142-1426 С 5-хлор-2-(2-пириднннлметнлсульфинил)-бепзоксазол (с),т.пл 91-93 С ММ-диметил-2(56.(5), т.пл. 1 в 9-19 о 5 с .н 2-(1 н-2 беиэимидазолнлсульфиннл)этнлн-метилбенэоламин (К), тпл.148 ь 149,5 С Ы,Ыднметнл-2-(1 Н 2-нафто-2,3-6 имидаволилсульфинилметил)-бензоламин (1), тдпл. 129 С (разлоЖВНИФ) 2-2-(1 Н-бензимидазолил 3-сульфинил-бензопамин (т), тыпп. 202-2 О 3 С дает усадку при 160 С 2(1 Н-2-бенэимндазолнлсульфинилме тил)-4-метоксн-М,М-дметнлбензоламин (п), т.пл. 13 о-131 с 2-(1 н 2-бенэимидааолилсульфннилметил)-Н-этилпропнлбензоламин (о), т.пл. 114 С 2-2-(4 морфолннил)-феннлметилсульфннил 1 Нбензимидазол (р).рагидро 1,6 диметилхинолин 8 илметил сулъфинип)-Нбензимидезол. у .1,23,4 Тетрагидро 11,6 диметил ХиЦ 0 пцн 8 кербоксальдегнд (а), Фосфорилхлорид (117 мл,1982 г,125 ммбль) добавляют по каплям крвстОРУ 12,З 4 дтетрагидро 1,6 ди метилхииолина (2,1 г, 10,3 ммоль) в СУХ 0 М.дИметилформамиде (7 мл) л атмосфере азота В условиях.перемешивалип при температуре ледяной бани. Реекционную смесь нагревают до 120 С(пе мгновение) и затем поддерживают при.8 ОС в течение 2 й. Смесь охлажа дают,.влнвают в разбавленный водный растворбикарбонатаиатрили 5 экстратируют этилацетатом (3 раза) Объедиленный органический слой промывают водой (3 раза), сушат надсулЬФатом натрия и выпаривают с получением целевого соединения в виде желтого масЛад 960 мг (492).тМРтспелтр(СВС 15) пожевал наличие альдегидепрн 10,06 чн/минодобдвляют порциями кпродукту арев течение 1 Омнн, Смесь ЦВРВМЕШН чают дополнительно В течение 20 мин,ацетатом (3 раза)д Объединенные органические слои промывают водой(2.ра за)сушат над сульфатом натрия идинения в виде вязкого бледножелтого месла 1,41 г(93) с 1 ш/22 Г(моно тмз проиьодндя) 263(штч,)248 (осповнрйпик),17273 ы 12,354 Тетрагндро 8-йлорметил пб-дшетиллинолина гидрохлорид (С) о Продукт Ь-(1,д гд 73 З ммоль) в сухом бензоле (10 мл) обрабатывают порциями тионилхлорида (О 8 мл,.13 г, 11 ммолв) при перемешивании вусловиях охлаждения холодной водя ной баней Температуру смеси доводятдо комнатной температуры (2 ч 7 изасмесь охлаждшот снова, обрабатывают эфирньм раствором хлористого водоро да-(2 мл) и вьтаривают досуха. Полу-. чающееся твердое коричневое вещество растирают с эфиром п фильтруют С получением целевого соединения в виде светлокоричпевого твердого оещества,1,73 г-(962). ш/г 209/11 (или), .175 (основной пик), 158, 145, 131, 119, 91.(100 мл) обрабатывают диметиланилит пом (15 мл) при Ос в услориях перемешивания Реакционную смесь затем перемешивают при комнатной температуре в течение 20 ч, Растворитель вьг паривают н продукт зкстрвгируютэтнл ацетатом(400 мл), промываютролным раствором бикарбоната натрия(100 мл) соляньм раствором (100 мл) и сушат над сульфатом магния. Растворитель вьшарирают н продукт отгоняют с помощью аппарата Ки 3 е 1 гцЬг (температу ра воздушного термостата 13770, 1,1 мм ртцст.) с получением 15,6 г целевого соединения ввиде бледнц желтого масла.(44,2 мл 1 М в эфире) добавляют пост тепеино к раствору продукта а (7,8 г) в сухом тетрагндрофуране (150 мл) при ОРС при перемешиваниив атмосфере озота. Смесь кипятят с обратным холодильником в течение 1,5 ч и затем резко охлаждают водой со льдом. Продукт экстрагируют этилацетатом(500 мл) проыввют соляным раствором-(2 й 100 мл), сушат над сульфатом магния и растворитель выпаривают.Продукт дистиллируют с помощью аппарата Кц 3 е 1 гиНг (темдрратура воздушного термостата О 5 С, 0,0 мм рт.ст.) с получением 5,65 г целевого соедине ния в виде бледножелтого масла. 3 Хлорметил 2 НН-диметиламино пиридина гидрохлорид (с)3 Тионилхлорид (3,25 ил) добавляют по каплям к перемешиваемому раствору продукта Ь(5,65) в духом дихлорме тане (100 мл) при 0 С в атмосфере азота. Температуру реакционной смеси доводят до комнатной н затем кипятят с обратим холодильником в течение 1,5.ч. Растворитель.выпарнвают, пров дукт отгоняют в виде аэеотропной смеси с топуолом и растирают с эфиром. Остаток, белое твердое веществопред ставляет собой целевое соединениесоединение (т.нл. 24126 С) по при меру 1 Ь . П р им е р 5. 2(Н 2 Вензимнда золилсульфииилметил)бензоламинд Ы 2 хлорметилфенил 2,д,6 триметилбензопсупъфонамнд (а). М(2 Гидроксиметилфенил)2,4 бт-триметнлбензолсульфонамид 06,0 цв сухом дихлорвтане (80.мл) обрабаты вают тионилхлорндом (115 мл) при КОМиатной температуре при перемешивании. Реакционную смесь перемешивают в течнне 5 ч, дополнительно добавляют тионилхлорид (0,1 мл) и перемешивание проделают в течение ночи. Затем реакционную смесь вливают в водУ Н органическийслой отделяют. Водный слой промывают дихлорметаном 0 РГ 3 ческие растворы объединяют, сушат Над сульфатом магния и растворитель выпаривают с получением 4,06 г ЦгЛеВ 0 Р 0 соединения в виде бледножелтого мас ла.(70 мл) в течение 3 ч. Реакционную смесь вливают в воду и-осажденный продукт отделяют фильтрованием, хорошо промывают водой н сушат с получением 4.39 г целевого продукта в виде темножелтото порошка, т.нл. 2022 озс.(4,83 г) обрабатывают при комнатной температуре метансульфоновой кислотой 129 мл) при перемешиваиннРеакционг ную смесь, окрашенную в красный цвет,перемешивают в течение 27 ч, медленно вливают внзбьггок водного раствора бикарбоната натрия и экстрагируют этилацетатом, который затем промывают солннвм раствором н высушивают. Растворитель выпаривают и остаток элюнруют из испарительной хроматографической колонки с использованием в качестве ЗЛЮЗНТЗ СМЕСЬ ДНХЛОРМВТЗНЗЗТНЛЗЦЕТЗТ(41) с получением д 3 г целевого продукта ввнде светпокоричневого твердого вещества с т.нл. 270 СПродукт с окисляют по приеру 1 Ь с получением после перекристаллизации из этанола целевого соединения в вирде пухообразиого бесцветного твердого вещества с т.нл. 177 С (разложение).оФторбензальдегид (8,68 г) и.Н метнлциклогексаламин (1,9 г) кипятят собратнм холодильником в димет тилформамиде (70 мл), содержащем кар бонат калия (14,49 г), при перемешивании в течение 5,5 ч. Охлажденную реакционную смесь вливают в разбавленную соляную кислоту н экстрагируют.вСНС 13. Водный слой отделяют,подщелачивают карбонатом калия и экстрагируют в СНС 13, который затем промывают водой, сушат и выпаривают спродукт а обрабатывают по примеру 56) с получением целевого соединения. ММД 219 (основной пик) 148.ПродуктЬ превращают в целевое соеч динение попримеру З, т.пл. 165 66 Сдинение по примеру 1 Ь,т.пл. 132 1 ззс.бП р и м е р- 7, По примеру 3 с нъч пользованием соответствующих исходных материалов может быть получен 2(1 Н/(70 Хтный водпыи раствор 15 г, 116 ммоль) в сухом хлористом метилене перемешивают в тоне азота на бане из смеси воды сольдом в течение 3 ч.По истечении 40 мин и 2 ч добавляют дополнительно 200 миллиграммовые апиквоты ванадилацетилацетоната. Далее смесЬ.выпаривают досуха в ро торном испарителе при комнатной температуре н подвергают немедленной быстрой хроматографической обработкеметанол. в качестве злюентаь Целевое соединение в количестве 700 мг (382) получают в виде бледно(м.1 Н) здзэ (м 1 н) 2,97 (м. 1 Н), 2,81(м. 1 Н) 2,25 (2.6 н) 1 Пр н ме р 9. Получение натриевык солей соединений указанной 064-Соединение указаннойобщей фор мулы растворяют в водном этанолезатем добавляют эквивалентный объем(75 г, 41,8 моль) в сн 1 с 1 (100 мл охлаждают льдом и обрабатывают 8001,(60 ммоль,7,1 е 3 д,38 мп).Получен ную смесь оставляют на сутки-при 25 С после чего отгоняют при 35 С в вакууме Остаток растворяют в сухом днметилформамиде(120 мл) и-добавляют к 5,бдиэтоксиг 1 Нбензимидазол 2днметилформамнде (25 мл) ив присутствии к,со (50 ммоль 69 г). Смесь перемешиваютд 8 ч при 2590, переносят в воду (800 мл) иекстрагнруют этил ацетатом-Экстракт пронываютводой и высушивают. Вытеснителъной хроматогра фней(Б 10 д, СНС 11 зтилапетат 41) получают указанное соединение в виде твердого вещества кремового цвета,т.пл 7882 С. . 2 ч(56 Диэтокси 1 Нбензиидазолч 2 илсульфннилметил)ММдиэтилбензамип (Ь).- б д2,95 г, 95). Реакцонную смесь перемешвают 2 ч при охлаждении льдом и эатемеще 2 ч при 25 С,промываютводным раствором ЫаН 03, затем воднпчараствором нансо, и водой. Реакционную смесь сушат, нспаряютиочищают вьгу теснительной хроматографией(З 104 р СН 1 С 11 этилацетат 31) с получением. вкачестве основного продукта смоло образного вещества(4,6 г)д Промыва нием эфиром получают заглавное сое динение в виде белого порошкаописанным способом наоснове соегут быть получены следующшесоедине ння 2(45 диметокси 51 н-бензимидае зол 2-илсульфиннлметил)МЫдиметил- бенэаминдчт.пл.1014102 С с размяг нением) 2(47-диметокси 1 н 4 бензнми
МПК / Метки
МПК: C07D 235/28
Метки: соединений, приемлемых, способ, производных, получения, сульфинильных, фармацевтически, гетероциклических, солей
Код ссылки
<a href="https://kz.patents.su/10-1863-sposob-polucheniya-sulfinilnyh-proizvodnyh-geterociklicheskih-soedinenijj-ili-ih-farmacevticheski-priemlemyh-solejj.html" rel="bookmark" title="База патентов Казахстана">Способ получения сульфинильных производных гетероциклических соединений или их фармацевтически приемлемых солей</a>
Способ получения правовращающих производных оксиизоиндолинила или их фармацевтически приемлемых солей

Номер патента: 1561
Опубликовано: 15.12.1994
Авторы: Джованни Карниель, Фабрицио Орци, Бруно Миорини, Джорджио Черони, Пьерлуиджи Гриджи
МПК: C07D 209/46
Метки: получения, оксиизоиндолинила, правовращающих, способ, солей, производных, фармацевтически, приемлемых
Формула / Реферат:
Изобретение касается гетероциклических веществ, в частности правовращающих производных оксииэоиндолина общей формулы I где Х = C6H4-пaрa-CHR-C(O)-O-R1 ; R=C1-С4-алкил; R=H или С1-С4-алкил, или их фармацевтически приемлемых солей, обладающих обезболивающей и противовоспалительной активностью, что может быть использовано в медицине. Цель изобретения - повышение выхода целевого продукта. Синтез ведут реакцией офталевого ангидрида с правовращающим...
Способ получения производных 2-тиенилоксиуксусной кислоты или их фармацевтически приемлемых солей

Номер патента: 1567
Опубликовано: 15.12.1994
Авторы: Дитер Биндер, Франц Ровенсцки, Хуберт Петер Фербер
МПК: A61K 31/38, C07D 333/26
Метки: фармацевтически, производных, способ, кислоты, 2-тиенилоксиуксусной, приемлемых, солей, получения
Формула / Реферат:
Изобретение касается производных 2-тиенилоксиуксусной кислоты, в частности получения 5-[2-(бензолсульфониламино)этил]- или 5-[2-(4-хлорбензолсульфониламино) этил]-2-тиенилоксиуксусных кислот, которые могут быть использованы в медицине для лечения тромботических заболевании. Цель - создание новых более активных веществ указанного класса. Синтез ведут окислением, например, амида N-[2-[2-(5-(2-гидрокси)-этокси)-тиенил]-этил]-бензолсульфоновой...
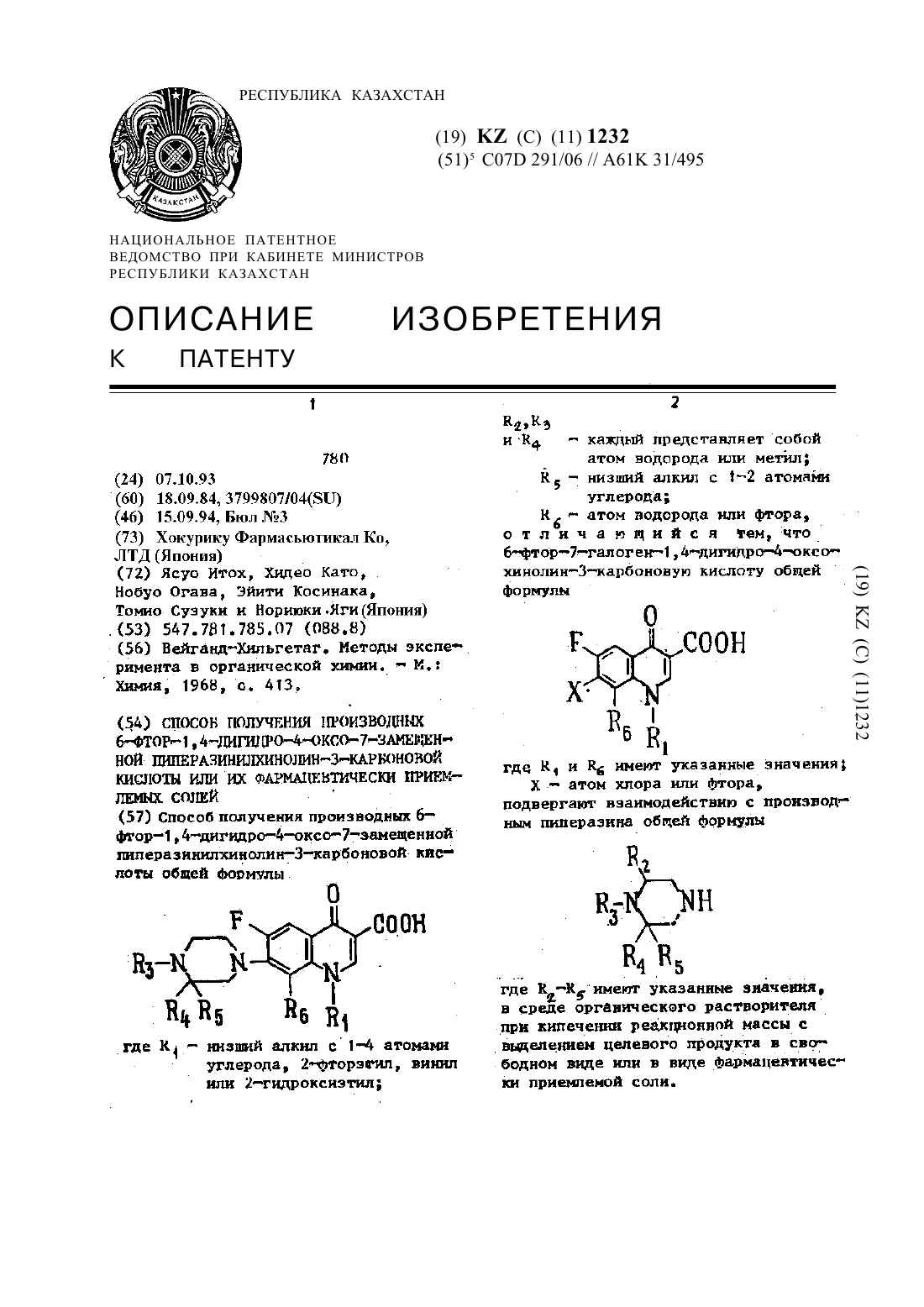
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты или их фармацевтически приемлемых солей
Номер патента: 1232
Опубликовано: 15.09.1994
Авторы: Нобуо Огава, Ясуо Итох, Эйити Косинака, Нориюки Яги, Хидео Като, Томио Сузуки
МПК: A61K 31/495, C07D 401/02
Метки: пиперазинилхинолин-3-карбоновой, кислоты, солей, способ, фармацевтически, 6-фтор-1,4-дигидро-4-оксо-7-замещенной, получения, производных, приемлемых
Формула / Реферат:
Способ получения производных 6-фтор-1,4-дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты общей формулыгде R1 - низший алкил с 1-4 атомами углерода, 2-фторэnил, винил или 2-гидроксиэтил; R2, R3 и R4 - каждый представляет собой атом водорода или метил; R5 - низший алкил с 1-2 атомами углерода; R6 - атом водорода или фтора, отличающийся тем, что 6-фтор-7-галоген-1,4-дигидро-4-оксо-хинолин-3-карбоновую кислоту общей формулыгде R1...
Способ получения замещенных аминодикислот, их рацематов или оптических изомеров, или их фармацевтически приемлемых солей

Номер патента: 1223
Опубликовано: 15.09.1994
Авторы: Жорж Ремон, Мишель Лоби, Мишель Винсен
МПК: A61K 31/47, C07D 217/26
Метки: получения, замещенных, приемлемых, фармацевтически, аминодикислот, рацематов, изомеров, солей, оптических, способ
Формула / Реферат:
Способ получения замещенных аминодикислот общей формулы 1 где А - бензольный цикл, n=1; А - насыщенный цикл n=0 или 1; R1 - низшая алкильная группа с 1-4 атомами углерода, содержащая аминогруппу; R2 - атом водорода или алкильная группа с 1-4 атомами углерода, R3 - линейная или разветвленная алкильная группа с 1-8 атомами углерода, моно- или ди- циклоалкил-алкильная группа или фенил-алкильная группа, содержащая в целом до 9 атомов углерода или...
Способ получения производных 13-галоидмилбемицина или их солей, или их сложных эфиров

Номер патента: 1235
Опубликовано: 15.09.1994
Авторы: Кацуо Сато, Акира Нисида, Бруно Фрай, Энтони О" Салливан, Норитоси Китано, Тосиаки Янаи
МПК: A01N 13/02, C07D 493/22
Метки: солей, сложных, получения, способ, 13-галоидмилбемицина, производных, эфиров
Формула / Реферат:
Изобретение относится к гетероциклическим соединениям, в частности к получению производных 13-галоидмилбемицина фор-лы I лы =N-OR2, где R2-Н, С1-С6 - алкил, или их солей, или их сложных эфиров, которые проявляют антигельминтную, акарицидную и инсектицидную активность.Цель - разработка способа получения новых более активных соединений. Получение их ведут галогенированием соединения фор-лы I, где R1 указано выше, в положении 5- O или ОН и Н, а в...