Способ получения нуклеозида или его фармацевтически приемлемых солей








Формула / Реферат
Изобретение относится к нуклеозидам, в частности к получению нуклеозида формулы
(НOН2С)СН-О-СН(R)-C(F)2-(OH)CH, где R - основание одной иэ формул
-N-C(O)-...-NH-C(O)-C(R1)=CH
или
-N-C(O)-N=C(NH2)-CR1=CH,
где R1, - водород, метил; фтор или йод, или их фармацевтически приемлемых солей, обладающих противовирусной активностью. Цель изобретения -выявление новых более активных соединении. Получение их ведут взаимодействием соединения формулы
(J10H2C)CH-0-Q-С(F)2--(J2O)CH,
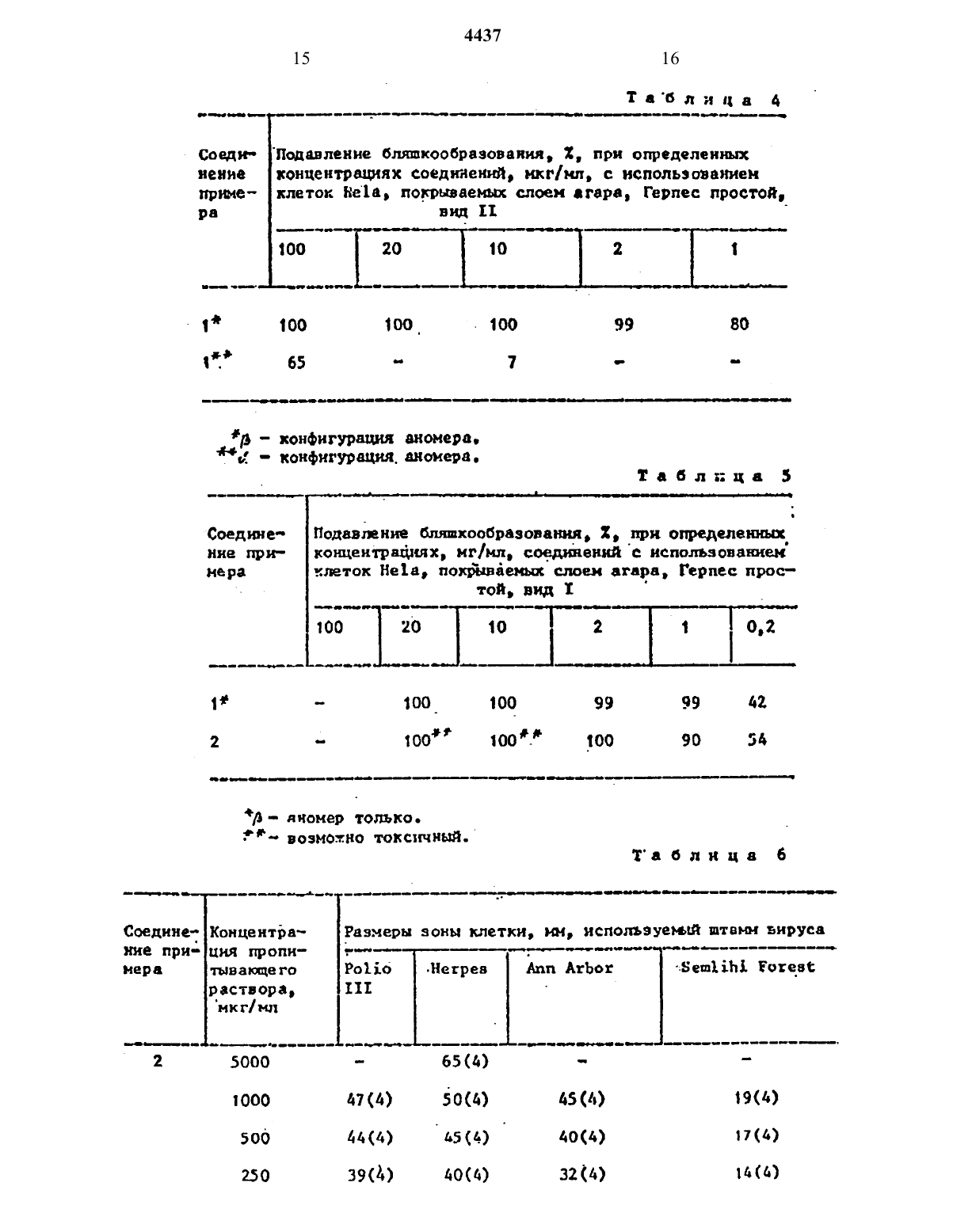
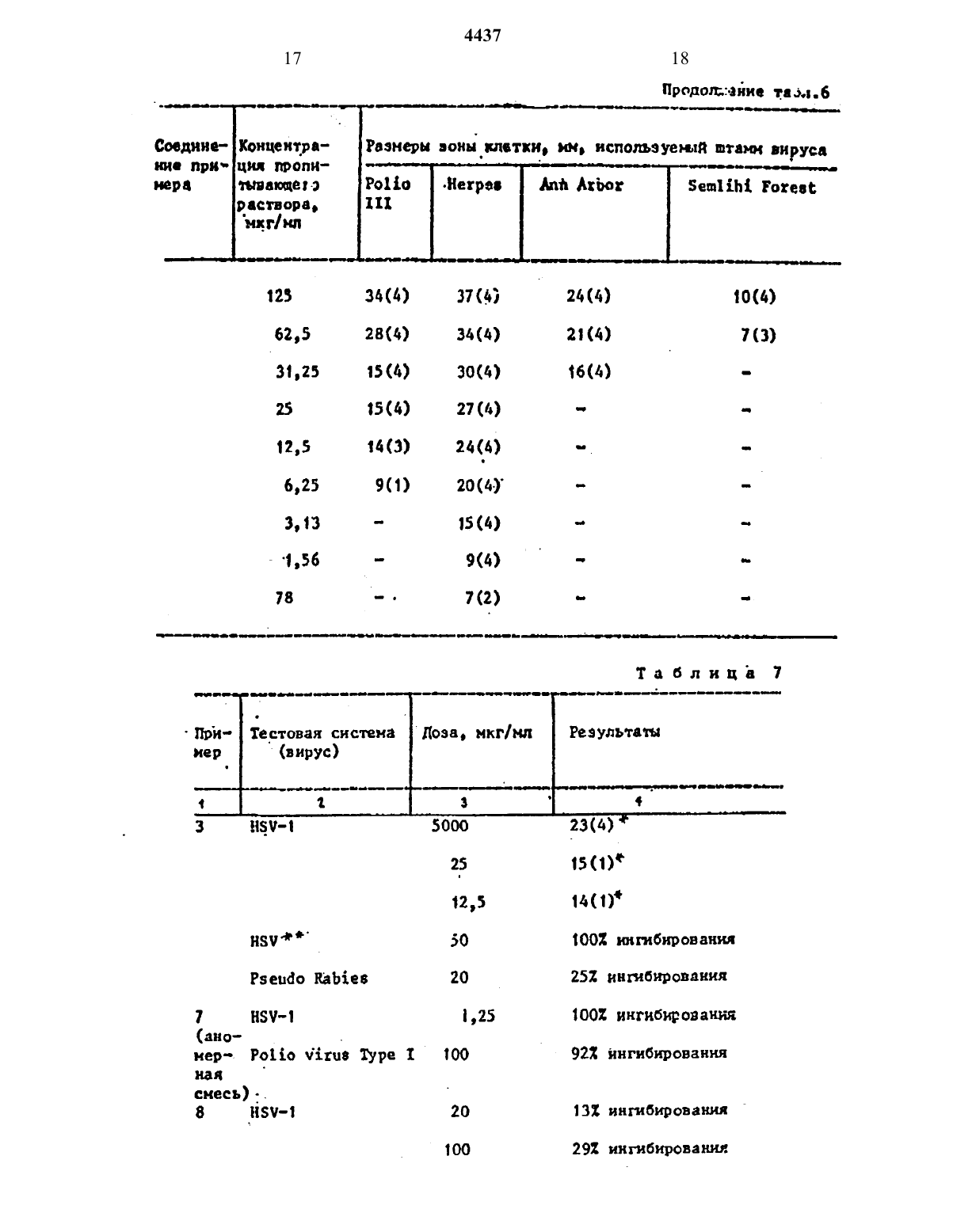
где Q - группа СНХ, где X - отщепляем ая группа; J1 и J2 - независимо гидроксизащитная группа, с защищенным основанием описанных формул. Процесс ведут в среде инертного растворителя, такого как 1,2-дихлорэтан или хлористый метилен, при температуре кипения реакционной смеси с последующим удалением защитных групп и выделением целевого продукта в свободном виде или в виде фармацевтически приемлемой соли, 7 табл.
Текст
изобретение относится к способу получения новых нуклеоэипоа обшей формулыгде К основание одной из формулйод, 7 или их фармацевтическн приемлемых солей, обладающих протишовнрУсН 0 д ЗКТХШПОСТЪЮоЦель изобретения - получение новых нукпеовидов, обладающих большей активностью, чем известный структурный аналог - видарабнп (Ара-А).В атмосфере азота к 2,59 г 3,5-бис(трет-бутипдиметнлсипокси)-1-нетансупьфоиплоксн-2 деэокон-2,2-дне фторорнбозы добавляют 1,60 г 5-метнл-24-бис-Стриметилокснлнлокси)пнридина и 45 мл сухого 1 д 2-дихпорэтана. Затем к этой смеси добавляют 1,45 г трифторнетансупьфоннлоксимеч тилсилана и перемешивают чистый раствор при кипячении с обратным холоднпьинкомв течение 2-3 ч. Затем-продукты реакции охлаждают до комнат-ной температуры, добавляют 1,35 мпгфильтруют, а фильтрат восстанавливают до половины своегообъена под 88 куумом н разбавляют равным образом дяхлорметана. Затем раствор промывают водным раствором бикарбоната натРИЯ И ПОСЛЕ зтогоиасьнщеиъъгмродшям раствором хлорида натрия и высушивют над безводным сульфатон натрия Раствор фильтруют, а филртрат насвеют безводным бромводородон. Резким ОННУЮ СМЕСЬ перемешивают В ТЕЧОНИЪ30 МИН Н ЗЗТЗМ СГУШЗЮТ ПОЛ ПЗКУУНОМ. ОСТВТОК РЗСТВОВЯЮТ В метаноле Н РЧСТвор выпаривают до сухости под вахуумом. Полученный остаток растворяют п воде и раствор дважды экстрагируют диэтиловым эфиром. Водный спой выпаривают. Остаток поглощают в этанолеИ ПОВТОРНО выпаривают до полного обезвоживания. Получают 1 г неочищенНОРО продукта, который хроматографируют на 30 г силикагеле Иое 1 ш (7010 меш), при эюироваини этилацетат том с выкодом 0,76 т требуемого продукте. Этот продукт дополнительно очищают переуристаплизацией из этилацетата с поручением 0,37 г белогоВ атмосфере азота К 5,0 г 3,5 бнс(третбутипщиметилсилокси)-1 ме таисупьфонилоксн 2-девоксн-2,2-днфторорибоэы В 1 МЛ СУХОГО 1,2-дихлор этана добавляют 5,68 г 6 истрнметнлсилипацетилцнтозина и затем 3,96 г трифторметансулъфоннлоксиметилснлана. Раствор кипнтлт с обратным холодильником в интервале 3-15 ч. Реакционную смесь 6 хлаждают до комнатной температуры, вводят 2 мл нетанола и полученную суспензию перемешивают в течение 30 мин. Остаток фильтруют, а фильтрат сгущают в вакууме до полного обезвоживания. ОстаТОК РЗСТВОРЯЮТ В МЕТНЛЗНХЛОРШЕЕ, На сыщают безводиы нвг и перемешивают при комнатной температуре в течение 5 мин. Полученную смесь сгущают до СУХОГО СОСТОЯНИЯ В ВЗКУУНЗ, ПОГЛОЩЗ ют метапольиым аммоннем И перемешивают в течене 15 мн при комнатной температуре. Затем раствор сгущают ДО ПОЛКОХО обезвоживания В ВБКУУНЕ,трижды разбавляют водой и водную ростворныую порцию вводят в колонку на силикагеле с обратной фазой, попучая 100 мг требуемого продукта при элюировании водой.-бис(трет-бутшщюетилсщокси)-1-мертансульфонилокси-2 дсзокси-2,2 тднфторорибозы в 1,35 мл сухого цг-ддхлорэтаие добавляют 2,08 г тристриметил силилт 5 йодцитозина и 1,11 г трифтормстаисульфонилоксметилсилана. Раствор кипятят с обрати холодильником в интервале 3-15 ч. Реакционную смесь Охлаждают до комнатной температуры,д 058 ВЛЛЮТ 5,0 мл метанола, полученную суспению перемешивают приблизительно в течение 30 ими. Остаток фильтру ют и фильтрат сгущают В вакууме.до СУхости. Остаток растворяют в метиленхлориде насыщают безводным Нвг и перенешишают при комнатной температуре в течение 45 мн. Смесь сгущают до сухого состояния в вакууме.0 статок трнщн насыщают водой, нейтрализуют ЗНСО 3 н разделяют из-колонне с сньликателен с обратной фазой (Ню НеОЪКЭН) с получением 26 мл требует мого продукта.В атмосфере азота х 1,1 г 3.5 бнс(третбутилдиетилсилоксн)-1-метансулъфонилокси-2 дезокон-2,2-дифторорибозы в 20 мл сухого дихлорэтана добавляют 2,88 г оис-трнметилснлип-5 чфторурацнла н 0,66 г трифторнетанн сулъфоалокситриметилсилаиа. Раствор охлаждают до комнатной температуры,добавляют 1,0 мл метанолаи полученНУю суспензиюперенешнвают примернов течение 30 ми. Остаток фильтруют и фильтрат сгущают в вакууме до сухо го состояния. Остаток растворяют в метилецхлориде насьщают безводным Нвг и перемешивают при комнатной температуре приблизительно в течение 45 ннн. Смесь сгущают до сухого состояния а вакууме. Остаток трижды нарсыщают Н 10, нейтрализуют НаНС 03 и разделяют на колонке с снликагелем с обратной фазой при эпюироьднии воДОЙ С ПОЛУЧЕНИЕ ТрЕбУЕМОЬО ЗСДУКТЗ.-5.4 г 3.5 бнс(третбутнлдпмстилдддКС)1 МетаНУЛЬФопилокси 2 дези окси 22 дифторорнбозы н 65,4 г 5-етип-2,дбис(тримстилснлилокси)пиримидиид соединяют и нагревают в атносфе ре азоте при перемешивании, при ООс 99 Ч 9 Не 1 Ч й ПРИ 150 Сп течение 1 ч. После этого смесь охлаждают до комиетной температуры, разбавляют 25 МЛ ЧОДН И 10 мл метанола. Сусцен. зю фильтруют через фильтрат со слоем кизелъгура и слой промъшают ацетоном. Соединенные фильтрата выпдрадЮТ ПОД ВЗКУУНОН и получают 5,3 г мвсч лянистого остатка. Остаток растворяют В 10 мл ацетона и вводят в Ь 5 сн колонку, набитую 80 г силикагеля. Проводят алюированме смесью дихлоре таиметанолтриэтиламии 1511.Первые 100 мл зпюеита отбрасывают,а следующие 300 ил выпаривают под вакуумом и получают 0,1 г сиропообрданого сырого продукта, который раствор рпют в 40 мл ацетона. Через раствор в течение 1 Ч барботируют хлористый водород, а. затем в течеииееще ч бврботнруют бромистый водород. После этого раствор выпаривают при 62 и получают ,4 г маслянистого темного продукта.Укаэаиньй продукт растворяют Л 10 мл теплой смеси днхлорнетаиуксусидя кислота 31 И вводят в колонку 6,5 см, набитую 45 г силикагеля. для первых 1000 мл в нечестно злюеита используют смесь дихлорметаиуксусиая кислота 31, посла чего В качестве элюента используют только укдусиую кислоту. Большая часть целевого продукта находится в фракциях между 1000 и 1400 мл, выходящих из колонки,как показывает тонкослойная хроматография на силикагеле с использованием снеси дихлорнетаннетаиоп 151. Эти Фракции объединяют и выпаривают под вакуумом, а остаток растворяют В 15 мл холодного ацетона и филътруют. Фильтрат выпаривают пол.вакуумОМ И получат масло, которое растворяют в 5 мл ацетона и хроматографирует на 20 г силикагеля с использованием смеси дихлорметанметаиол 151. Содержащие продУКТ ФРЗКЦНН 0 бЪ 8 дЯТэвыпаривают ПОД ВЗКУУНОН Н получают 300 мг полужидкого соединения. Этот продукт растворяют в 5 мл ацетона и фильтруют, фильтрат выпаривают под вакуумом и получают 230 мг светлокорнчневого лолутверлото соединения. Его растворяют в 10 мл носьпюнного водного раствора бикарбоната натрия и раствор экстрагируют дважды порциями по 15 мл диэтилового эфира. после этого водную фазу выпаривают ПОД ВЗКУУМОМ, ОСТЯТОК суспеидирупот в ацетоне и фильтруют.фнльтрат выпаривают под вакуумом и получают 10 мг целевого продукта в виде рыжевато-коричххевотр вязкото масла.К 80,0 г 3,5-бис(трет-бутилдиме. тилсиллокон)1 метансульфоннлоксм 2-дезокси-2,2 дифторорибоэы в атмосфере азота.прнбавллют 1,4 л свежеис ВВГНВННОГО КЛОРИСТОГО ме-тиленз И49,5 г 5 нетил 2 дбис(триметилсилил окси)примидниа. К этой смеси прибавляют 44,8 г трифторметансульфонил окситриметилсилана и реакционную смесь кипятят с осратнъм холодильнком приблизительно в течение 3,25 ч. Реакционную смесь перемешивают прилриблзнтельно в течение 30 мин,оса дишшееся твердое соединение собираютпод вакуумом при 45 с с получением,темного масла, которое растворяют В 500 мл хлористого метилена, насыщенного безводньм бромнстым вод 0 РОд 0 МРезультмрующую суспензию перемещи вают приблизительно в течение 3 Ч,после чего летучие соединения Уд 8 ПЯ ют под вакуумом при 45. Остаток растворяют в 100 мл 102 ного раствот ра бикарбоната натрия м 100 МЛ днэтилового эфира. Вдкьй слой отдепя ют.и концентрируют под вакуумом при 50 С и получают остаток, который триды растворяют с порциями по100 мл горячего этилацетата. 05 аии ческие слои объеднптот выпаривают под вакуумом при д 5 н получают ос таток, который растворяют п 50 мл воды. Этот раствор хроматографируют порциями по 10 мл на обращс ио-фа 3 овой колонке Пасете Ртер 500 С 9 с(трет-6 утип 5 иметилсилнлокси)2-метансульфонилокси 2 дезокеи 22-дифтерксилоэы прибавляют 23 г трие(триме тилсилил)цитозина и 300 мл хлористого метилена. К этой смеси прибавляют 10,84 гтрифторметансулъбоннлокси-. триетилсипана и смесь кипятят с обратим холодильником приблизительно в течение 16 Ч. Смесь охлаждают до комнатной температуры и 20 мл метанола прибавляют к смеси. Раствор анергично перемешивают приблизительно в течение 1 ч при комнатной температуре, и выпавшее в осадок твердое соединение собирают фильтрованием.100 мл поди и суспензию энергично перемешивают в течение 30 мин. Оргап пический слой отделяют и выпаривают досукого остатка при пониженном давлении, получают 11,2 г коричнепоч то масла. Остаток растворяют в 97 мл метанола, к раствору прибавляют 33 г катионообменной смолы В 10 каа А.с. 50 Х 8. Суспензи переманивают ЛРНПВ эительно в течение 16 ч при комнатной температуре и смолу собирают фильтрованием. смолу промъшают 50 мл метанопа и энергично перепахивают в растворе 100 мл метанола и 100 мл гидро окиси аммония. Эту операцию повторяют два раза, объединенные фильтры кони центрируют под вакуумом при 50 и получают 2,09 г желтого остатка. Ос таток суспеидируют в 25 мл воды и эпеьгньно перемешиают Ь течение15 ммн. нерастворимый осадок сооирач ют фильтрованием и получают 0,25 т соединения А. Фильтрат концентРНРУютр под вакуумом при 50 и полУчают30 мл 11 (объем/объем) раствора метанол/гидроокись аммония. Смолу снова фищтруют под вакуумом при 50 С и получают 0,14 г 1-(2-дезокси-2,2Вдатмосфере азота раствор 1,86 г 3,5 бис(трет-бутилднметилсилилокси) ч 2-метансутафонилокснт 2-де воксмт 2 , 2 чхифторорибовы, 1,87 г трис-гриметилсилмлцитоэина и 1,34 г трифторметансулъфоннлокситриметилснлвна в 37 улсухого хлористого метилеиа кипятят с обратным холодильником втеченне ночи. Реакционную смесь охлаждают до комнатной температуры нв нее прибавляют 1 мл метанола. выпавшее в осадок твердое.соеднненне собирают фильтрованием и фильтрат вы- паривают под вакуумом при 65. Остаток растворпют приблизительно в 20 мл воды к этот раствор концентрирушат приблизительно до половины его первоначального объема выпариванием под вакуумом при 50. Остаток не- ч сколько рая растирают с порциями по 10 мл теплого ацетона. Органические экстракты объединяют, вьшаръшаютпод вакуумом при д 5 ии получат 3,67 гжелтого масла. Это соединение раство ряют в 15 млсмесн метанол/вода(объем/объем, 21) н перемешиают в течение ночи с 5 г 510 ад АС 501438 Суспенаюо иасьщат безводным аммиаком и перемешивают приблизъпельяо втечение 10 мин. Смопу собирают вакуумном фильтрованием н суспепдпру ют в 30 мл снеси метанол(анмиак (1 ПО ОбЪМУ)о Суспензии перемешивают приблизительно в течение 10 мин. Смолу собирают фильтрованием, основные фильтраты объединяют, концентрируют под вакуумом лри 50 и получают 1,5 г оранжевого масла. Масло растпориют в 10 мл воды, этот раствор хроматогртфируют порциями по 2 мл препаративной (50 см) колонке с обращенной фазой Ипнстап рас 1 в 11 0 П 33 с использованием воды в качестве элюеита и получакт 0,07 г 1-(5-метил 2 оксо-дамино-1 Н-лпримидин 1-ил)-2-дезоксп-2,2 дНФТ 0 рорибозы. ЯМР ССВЭОВ, 90 МГЦ), д 1,94 (сннгпет,ЗН), 3,53-4,62 (мультиплет, ДН) 6,75Противовирусиая активность соедннений в соответствии с изобретением показана проводным испытаниями йп чйсто, которые аюполнялись по следующей методике.почечные клетки африканской зеленой обезьяны (ВЗС-1 или клетки Не выращивают В 25 см флаконе производства фирмы Ра 1 соп при 3770 в среде.сыворотки бычьего эмбриона (В 3 С 1) пеиицилпин (150 МЕ/мл) и стрептоми цин (150 мкг/мл). Когда образовались спивающиеся монослоя, ростовую культурную срецу сливают и 0,3 мл соответствующего разбавлении указанного вируса добавляют в каждую чашку. После адсорбции втечение 1 ч при комнатной температуре вирус, который внедрялся в клеточный слой, был покрыт культуральноп средой, содержащей 1 ч ионагра Ю 2 н 1 ч среды 199 с удвоенной концентрацией, включающей сыворотку телячьего эмбриона. 51, пеннцкллмн, стрептомицин, а также исследуемое соединение ПРИ УКЗЗЗННЫХ концентрациях, выраженных В ННКР 0 граммах на миллилитр (мкг/Мл). Фпа дон, 3 котором не было нсптуенк соединении, служил как контрольныйОсновиые растворы испытуемого соединения выполнены в диметилсулъфоксиде
МПК / Метки
МПК: C07H 19/06, A61K 31/70
Метки: способ, нуклеозида, фармацевтически, солей, приемлемых, получения
Код ссылки
<a href="https://kz.patents.su/10-4437-sposob-polucheniya-nukleozida-ili-ego-farmacevticheski-priemlemyh-solejj.html" rel="bookmark" title="База патентов Казахстана">Способ получения нуклеозида или его фармацевтически приемлемых солей</a>
Способ получения замещенных аминодикислот, их рацематов или оптических изомеров, или их фармацевтически приемлемых солей

Номер патента: 1223
Опубликовано: 15.09.1994
Авторы: Мишель Винсен, Мишель Лоби, Жорж Ремон
МПК: A61K 31/47, C07D 217/26
Метки: оптических, способ, рацематов, солей, аминодикислот, получения, приемлемых, фармацевтически, изомеров, замещенных
Формула / Реферат:
Способ получения замещенных аминодикислот общей формулы 1 где А - бензольный цикл, n=1; А - насыщенный цикл n=0 или 1; R1 - низшая алкильная группа с 1-4 атомами углерода, содержащая аминогруппу; R2 - атом водорода или алкильная группа с 1-4 атомами углерода, R3 - линейная или разветвленная алкильная группа с 1-8 атомами углерода, моно- или ди- циклоалкил-алкильная группа или фенил-алкильная группа, содержащая в целом до 9 атомов углерода или...
Способ получения производных 2-тиенилоксиуксусной кислоты или их фармацевтически приемлемых солей

Номер патента: 1567
Опубликовано: 15.12.1994
Авторы: Дитер Биндер, Франц Ровенсцки, Хуберт Петер Фербер
МПК: A61K 31/38, C07D 333/26
Метки: приемлемых, 2-тиенилоксиуксусной, кислоты, способ, солей, фармацевтически, производных, получения
Формула / Реферат:
Изобретение касается производных 2-тиенилоксиуксусной кислоты, в частности получения 5-[2-(бензолсульфониламино)этил]- или 5-[2-(4-хлорбензолсульфониламино) этил]-2-тиенилоксиуксусных кислот, которые могут быть использованы в медицине для лечения тромботических заболевании. Цель - создание новых более активных веществ указанного класса. Синтез ведут окислением, например, амида N-[2-[2-(5-(2-гидрокси)-этокси)-тиенил]-этил]-бензолсульфоновой...
Способ получения сульфинильных производных гетероциклических соединений или их фармацевтически приемлемых солей

Номер патента: 1863
Опубликовано: 15.03.1995
Авторы: Джон Льюис Сучитский, Девид Кокс, Энтони Говард Инголл
МПК: C07D 235/28
Метки: производных, солей, приемлемых, способ, соединений, гетероциклических, сульфинильных, фармацевтически, получения
Формула / Реферат:
Изобретение касается гетероциклических соединений, в частности их сульфиннльных производных общей формулы M-S(0)-Rc (I), где М - группа формулы (II), а Rс - группа формулы -(СR10 R17)у - (CR18R19)z-Rx, где Y и Z (равные или разные) - 0 или 1, или 2; R16,R17 и R19-Н и Rх- группа формул (III), (IV) или (V):и, когда (Y+Z) не = 0, Rx может быть NR9R10 R2, R3, и R4 (равные или разные) - II, галоген, алкокси, алкил или -N(R)2 , или смежная пара R1,...
Способ получения ( 2 – морфолинофенил) гуанидинов или их фармацевтически приемлемых солей

Номер патента: 2848
Опубликовано: 15.12.1995
Автор: Баласубраманьян Гопалан
МПК: C07D 295/135, A61K 31/535
Метки: морфолинофенил, фармацевтически, солей, получения, гуанидинов, способ, приемлемых
Формула / Реферат:
Использование: в качестве противодиабетических средств в медицине. Сущность изобретения: продукт (2-морфолмнофенил)-гуанидины общей формулы где NR1R2 - метиламино, диметиламино, этиламино, метилэтиламино, бутиламино, аллилметиламино, этилметоксиэтиламино, диаллиламино, N'-метилпиперазинил, диметилморфолино, метилпиперидинил, морфолино или тиаморфолино и R3-Н, F, CI. СН3 или СН3О, или их соли. Реагент 1: HNR1R2. Реагент 2:...
Способ получения правовращающих производных оксиизоиндолинила или их фармацевтически приемлемых солей

Номер патента: 1561
Опубликовано: 15.12.1994
Авторы: Пьерлуиджи Гриджи, Джованни Карниель, Джорджио Черони, Фабрицио Орци, Бруно Миорини
МПК: C07D 209/46
Метки: фармацевтически, оксиизоиндолинила, получения, производных, приемлемых, способ, правовращающих, солей
Формула / Реферат:
Изобретение касается гетероциклических веществ, в частности правовращающих производных оксииэоиндолина общей формулы I где Х = C6H4-пaрa-CHR-C(O)-O-R1 ; R=C1-С4-алкил; R=H или С1-С4-алкил, или их фармацевтически приемлемых солей, обладающих обезболивающей и противовоспалительной активностью, что может быть использовано в медицине. Цель изобретения - повышение выхода целевого продукта. Синтез ведут реакцией офталевого ангидрида с правовращающим...
Предыдущий патент: 2-метил-10-(4-метил-1-пиперазинил)-4Н-тиено [2,3-В] [1,5]-бензодиазепин или его кислотно-аддитивная соль, 4-амино-2-метил-10Н-тиено (2,3 – В)( 1,5) бензодиазепин
Следующий патент: Эфиры эстрамастина или их фармацевтически приемлемые соли и способ их получения
Случайный патент: Ароматические гетероциклические соединения, способ их получения, фармацевтическая композиция на их основе и способы лечения