Способ получения антрациклинонов
Номер патента: 3938
Опубликовано: 16.09.1996
Авторы: Сильвиа де Бернардинис, Илариа Кандиани, Валтер Кабри, Франко Франкаланчи



Формула / Реферат
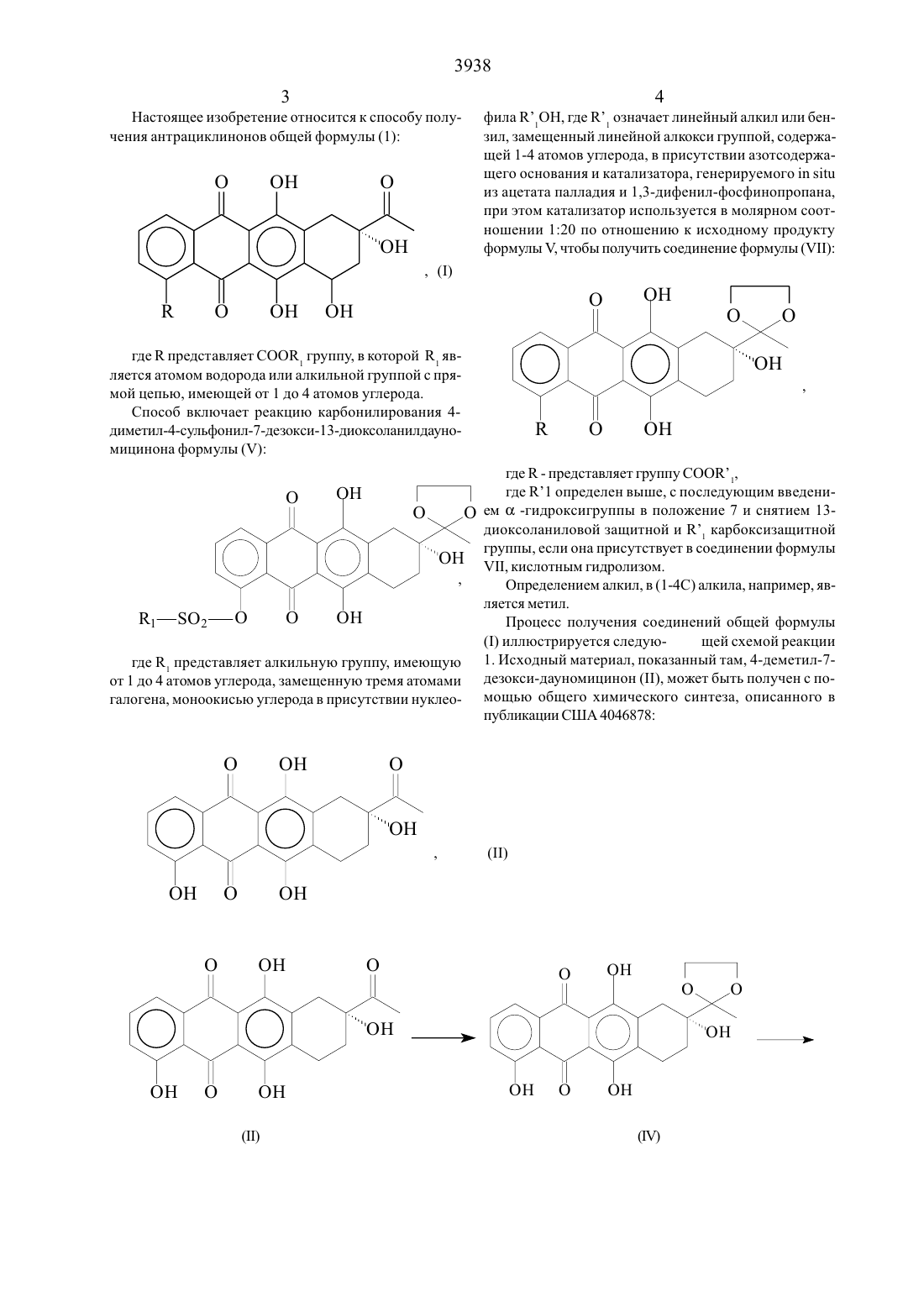
Настоящее изобретение относится к способу получения антрациклинонов общей формулы (1):
, (I)
где R представляет СООR1 группу, в которой R1 является атомом водорода или алкильной группой с прямой цепью, имеющей от 1 до 4 атомов углерода, карбонилированием 4-диметил-4-сульфонил-7-дезокси-13-диоксоланилдауномицинона. Настоящее изобретение позволяет синтезировать целевые соединения общей формулы (1) с высоким выходом и без стадий оптического расщепления или разделения. Соединения (1) являются промежуточными продуктами при получении противоопухолевых антрициклиновых гликозидов.
Текст
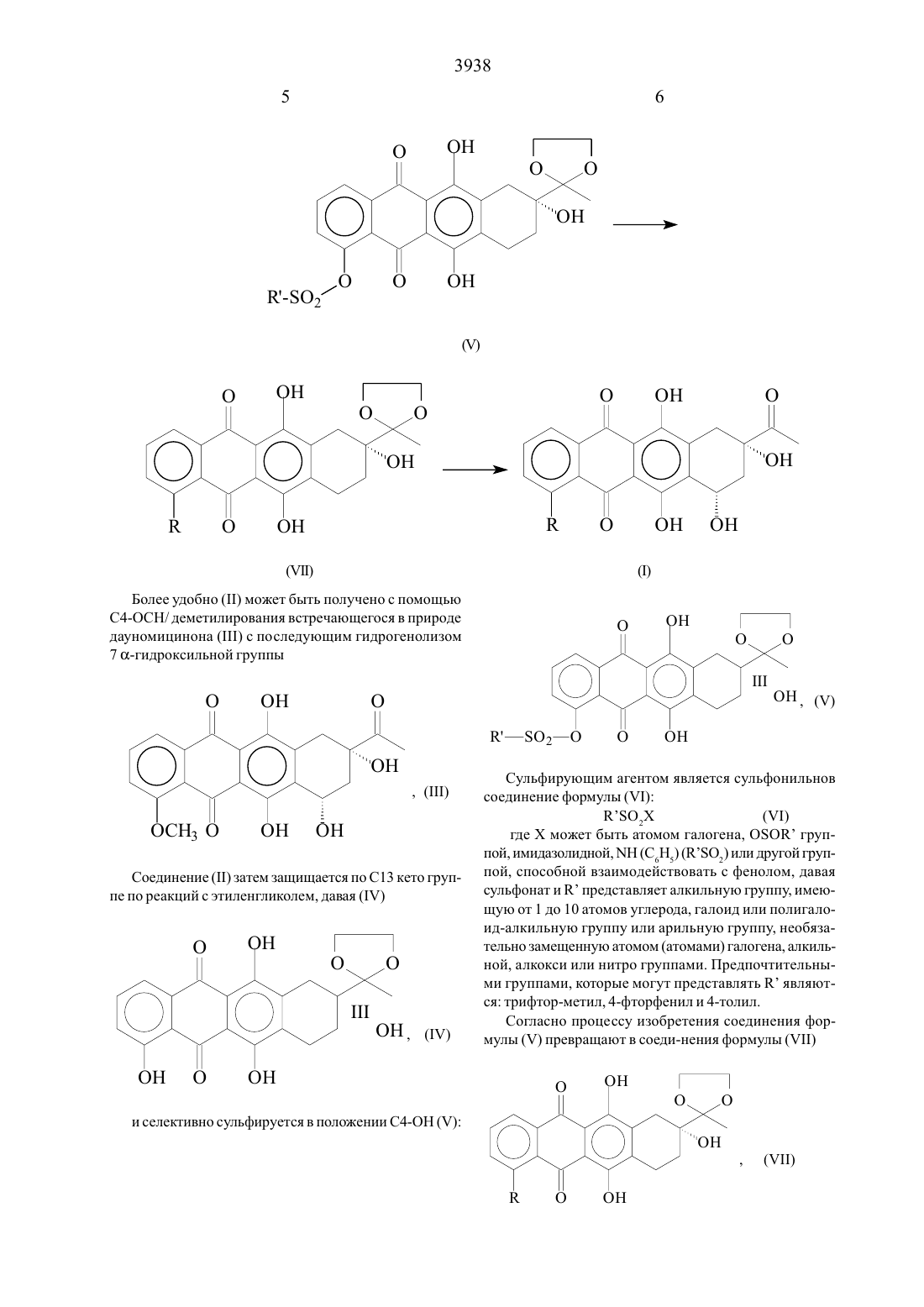
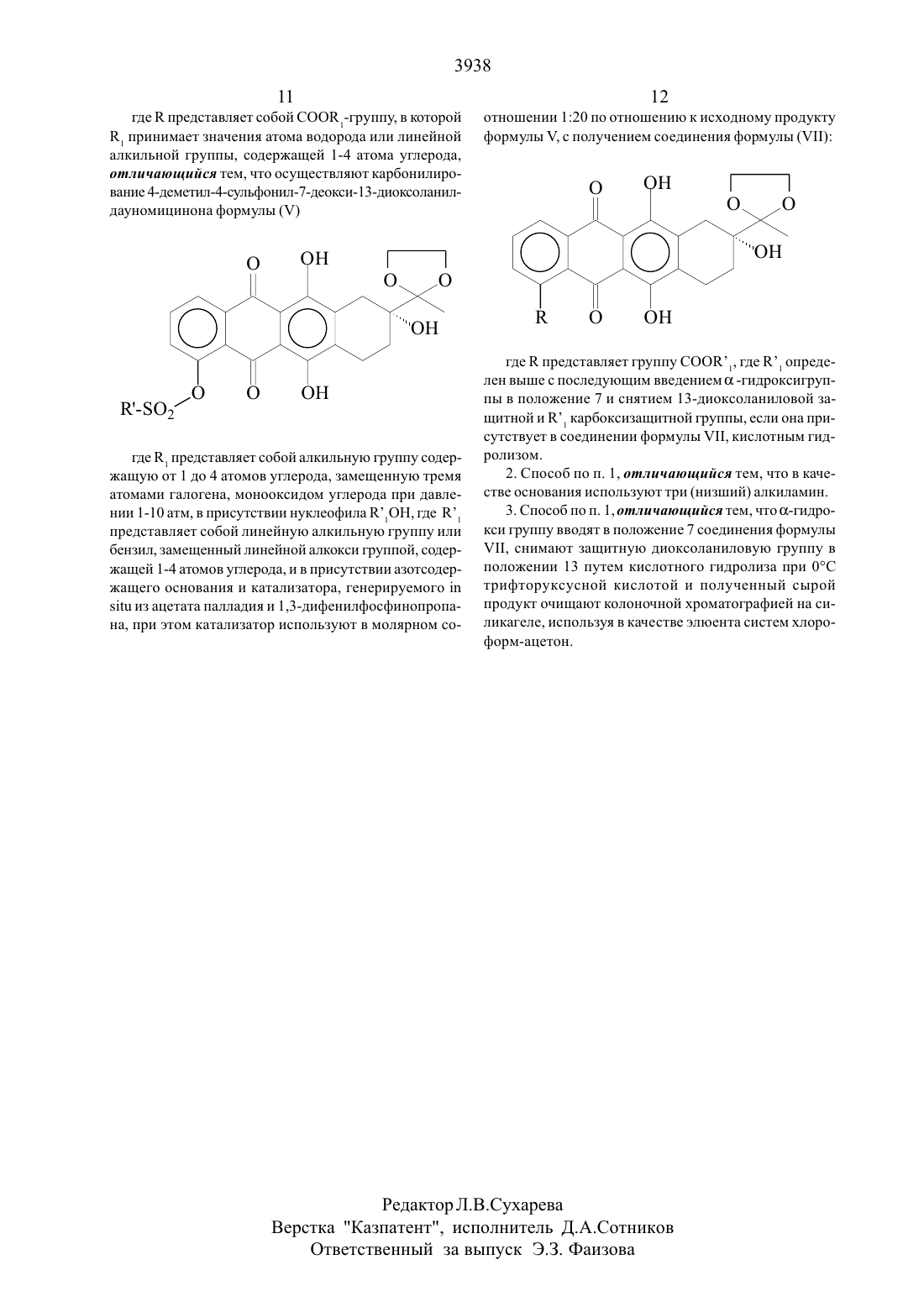
Настоящее изобретение относится к способу получения антрациклинонов общей формулы (1)где К представляет СООК группу, в которой К является атомом водорода или алкильнои группои с прямой Цепью, имеющей от 1 до 4 атомов углерода.Способ включает реакцию карбонилирования 4 диметил-4-сульфонил-7-дезокси- 1 3 -диокс оланилдауномицинона формулы (У)где К представляет алкильную группу, имеющую от 1 до 4 атомов углерода, замещенную тремя атомами галогена, моноокисью углерода в присутствии нуклео 44фила К 1 ОН, где К означает линейный алкил или бензил, замеЩеннь 1 и линеинои алкокси группои, содержаЩей 1-4 атомов углерода, в присутствии азотсодержаЩего основания и катализатора, генерируемого 111 5111 из ацетата палладия и 13-дифенил-фосфинопропана при этом катализатор используется в молярном соотношении 120 по отношению к исходному продукту формулы И чтобы получить соединение формулы (УП)где К - представляет группу СООК 1, где К 1 определен выше, с последующим введением (х -гидроксигруппы в положение 7 и снятием 13 диоксоланиловой защитной и К карбоксизаЩитной группы, если она присутствует в соединении формулы УП, кислотным гидролизом.Процесс получения соединений общей формулы(1) иллюстрируется следую- Щей схемой реакции 1. Исходный материал, показанный там, 4-деметил-7 дезокси-дауномицинон (П), может быть получен с по мощью общего химического синтеза, описанного в публикации США 4046878Более удобно (11) может быть получено с помощью С 4-ОСН/ деметилирования встречающегося в природе дауномицинона (111) с по следующим гидрогенолизом 7 ос-гидроксильной группыСоединение (П) затем защищается по С 13 кето группе По реакций с этиленгликолем, давая (П)СулЬфируюЩим агентом является сульфонильнов соединение формулы (/1) К О,Х (/1) где Х может быть атомом галогена, ОЗОН группой, имидазолидной, МН (С БНЗ) (КО 2) или Другой группой, способной взаимодействовать с фенолом, давая сульфонат И К представляет алкильную группу имеюЩую от 1 до 10 атомов углерода, галоид или полигалоид-алкильную группу или арильную группу, необязательно замещенную атомом (атомами) галогена, алкильной, алкокси или нитро группами. Предпочтительными группами, которые могут представлять К являются трифтор-метил, 4-фторфенил и 4-толил. Согласно процессу изобретения соединения формулы (У) превращают в соеди-нения формулы (УП)где К означает группу СООК 1 с помощью карбонилирования соединения (У) монооксидом углерода при Давления 1-10 атм в присутствии нуклеофила К 1 ОН, где Кн означает линейный С 1-С 4 алкил или бензил, замещеннь 1 й линейной С 1-С 4 алкокси группы, и азотсодержащего основания И катализатора, генерируемого из ацетата палладия И 1,3-дифенилфосфинопропана, где катализатор используется в молярном соотношении 1 20 по отношению к исходному продукту У. Подходящими основаниями являются триалкиламины. Температура реакции в типичном случае составляет от 0 до 150 С.7-0 с-гидроксильная группа затем вводится в соединение формулы (ЧП), а 13-диоксоланиловая и К карбоксизащитная группа, если присутствует, удаляется,давая конечные соединения формулы (1). Введение 7 ос-гидроксильной группы может осуществляться по способу, описанному авторами С.М. 91/0113 И др., Сап 1. С 11 еш. 5 1,446 (1973) бромированием соединений (УП) в С 7 положении И гидролизом 7-бром И 13-кетальной групп, давая соединения формулы (1).Типично ос-гидрокси группа вводится в 7-положение соединения формулы (ЧП), 13-диоксоланИл-заЩищающая группа удаляется с помощью кислотного гидролиза при 0 С трифторуксусной кислотой, и полученный неочищенный или сырой продукт очищается с помощью хроматографии на силикагельной колонке с использованием системы элюента хлороформ-ацетон. Для К, представляющего СООКР системой может быть хлороформ-ацетон (955 объем/объем).Хотя использованием катализа с помощью переходных метадшов для карбонилирования арилсульфанатом,было известно уже в течение ряда лет, это является новым в химии антрациклинов. Процесс настоящего изобретения, исходящий из обычного сульфоната формулы (У), дает возможность синтезировать несколько цепных промежуточных продуктов общей формулы (1),которые в противном случае доступны только с помощью индивидуального синтеза. Кроме того, когда исходный материал (11) получается из встречающегося в природе дауномицинона (У), настоящее изобретение позволяет синтезировать Целевые молекулы общей формулы (1) с высоким выходом и без стадий оптического расщепления или разделения. Соединения (1) являются промежуточными продуктами при получении противоопухолевых антрициклиновых гликозидов.Настоящее изобретение теперь будет описано более полно с помощью следующих ниже примеров, которые даются лишь с целью иллюстрации и никоим образом не предназначены для ограничения настоящего изобретения.К суспензии 13 г 35,3 ммоля) 4-Деметил-7-дезоксИдауномицинона в 400 мл бензола добавлялись 30 мл этиленгликоля и 0,3 г пара-толуол-сульфокислоты. Реакционная смесь нагревалась с обратным холодильником с азеотропным удалением воды в течение приблизительно 6 часов, затем охлаждалась до комнатной тем 8пературы. Твердое вещество выделялось с помощью фильтрования и промывалось водой И этанолом, давая после сушки 13,1 г соединения (1/). Данные анализа НР 2 С показали, что продукт был 98,6 чистоты. Анализ НР 2 С (жидкостная хроматография вь 1 сокого давления или высокой разрешающей способности) Колонка МЕРК КР 18 /7 мкм /250 х 4,2 мм/ Подвижная фаза А - 0,01 М гептасульфонат натрия (0,02 М фосфор ная кислота) 6 Ацетонитрил 4 В- метанол 7 Ацетонитрил 3Детектор УФ при 254 нм.ТСХ на Кизельгельной пластине Г 254 /Мерк/ с использованием смеси хлороформ / ацетое /9/ 1 по объему/ 3150,62К раствору в пиридине (1 10 мл/ 1 ,1 г/2,7 ммоля,) соединения (1/), 2,3 мл (13,2 ммоля) диизопропИлэтиламина и 0,33 г (2,7 ммоля) 4-диметиламинопиридина,охлажденному при 0 С, добавлялось 1,4 мл /8,3 ммоля/ трифторметансульфонилангидрида, и реакционная смесь перемещивалась в течение 1 часа при комнатной температуре. Реакционная смесь затем охлаждалась до 0 С, и к ней добавлялось 500 мл метиленхлорида и 300 мл 10 соляной кислоты. Органическая фаза промывалась водой, сушилась над сульфатом натрия,И растворитель выпаривался при пониженном давлении, давая твердое вещество, которое нагревалось с обратным холодильником в течение 15 минут в метаноле /35 мл/ и фильтровалось и получалось 0,95 г /65 от 1//СоединеНия/Д КСР 3/. /НР 2 С 94, условия, как описаны в примере 1/.ТСХ на Кизельгельной пластине Г 254 /Мерк/ с Использованием смеси хлоро-форм - ацетон /9/ 1 по объему/ КТ 0,58.К раствору 2 г соединения /А КСР 3/, /3,6 ммоля/ в 50 мл диоксана в атмосфере окиси углерода последовательно добавлялись 1 мл триэтиламина, 3 мл метанола,74 мг 1,3-дифенилфосфинопропана /0,178 ммоля/ И 40 мг ацетата палладия /0, 178 ммоля/. Реакционная смесь перемешивалась при 6 ОС до тех пор, пока не прекраЩалось Поглощение СО, затем охлаждалась до 0 С, подкислялась 10 соляной кислотой и экстрагировалась метиленхлоридом. Органическая фаза вь 1 паривалась досуха, оставляя 1,44 г / 88,1 / сырого 4-диметокси-4 метокси-карбонил-7-дезокси- 1 3 -диоксоланил-дауномицинона//11,КСООСН 3/, /НР 2 С 95,1 /.ТСХ на Кизельгельной пластине -Р 254 /Мерк/ с использованием смеси хлороформ /ацетон/ 91 по объему/ КГ 0,54.Соединение, описанное выше //Н, Р СООСН 3/,/ 1,4 г 3,08 ммоля/ растворялось в 200 мл четыреххлористого углерода, нагревалось при температуре Дефлегмации, и к раствору добавлялся 2,2-азо-изобутиронитрил /0,68 г/ и 200 мл воды. К реакционной смеси, перемешиваемой энергично, добавлялось по каплям на протяжении 30 минут 5,9 мл 0,6 М раствора брома в четыреххлористом углероде. Спустя 1 час, смесь охлаждалась, и органическая фаза промывалась водой и экстрагировалась 1 норм. гидроокисью натрия. Величина рН водного раствора щелочи доводилась до 8,2 с помощью 2 норм соляной кислоты, и смесь экстрагировалась метиленхлоридом. Раствор сушился над сульфатом натрия, и растворитель упаривался в вакууме. Остаток растворялся в 37 мл трифторуксусной кислоты и 4 мл воды при 0 С и перемешивался в течение 1 часа, реакционная смесь затем разбавлялась 60 мл воды и экстрагировалась метиленхлоридом. Органическая фаза промывалась водным бикарбонатом натрия и водой и сушилась над сульфатом натрия. Растворитель удалялся в вакууме, и остаток хроматографировался на силикагеле /хлороформ-ацетон 9515 по объему в качестве элюента/, и получалось 0,71 г /54,1 из УП,РСООСН 3/соединения/ 1,КСООСН 3/, /НР 2 С 98,7 /.ТСХ на Кизельгельной пластине Р 254 /Мерк/ с использованием хлороформ ацетон /91 по обьему/ КОАО/9,8 г, 72 ммоля/ использовался вместо метанола. После прекращения поглощения СО реакционная смесь обрабатывалась, как описано в примере 3, давая 1,65 г сырого 4-деметокси-4-/4-метоксибензил/карбонил-7 Дезокси- 13-диоксоланил-дауномицинона / УП,КСООСН 2/ С ЬН 4/ОСН 3/ , /Н Р 2 С 96,3/.ТСХ на Кизельгельной пластине Р 254 /Мерк/ с использованием смеси хлороформ/ацетон /91 по объему/ 0,55.1. Способ получения антрациклинонов формулы 1
МПК / Метки
МПК: C07C 50/36
Метки: антрациклинонов, способ, получения
Код ссылки
<a href="https://kz.patents.su/6-3938-sposob-polucheniya-antraciklinonov.html" rel="bookmark" title="База патентов Казахстана">Способ получения антрациклинонов</a>
Способ получения оптически активного ( + ) 4- деметоксидауномицинона

Номер патента: 3471
Опубликовано: 10.06.1996
Авторы: Уолтер Кабри, Сильвиа де Бернардинис, Серджио Пенко, Франко Франкаланчи
МПК: C07C 50/36
Метки: получения, активного, деметоксидауномицинона, оптически, способ
Формула / Реферат:
Использование: в медицине в качестве антибиотика или противоопухолевого агента. Сущность изобретения: продукт - оптически активный (+) деметоксидауномицион. Реагент 1:4-деметил-13- Реагент 2: сульфонилхлорид ф-лы R-SO2-CI, где R - трифторметил. Условия реакции:, N-диизопропилэтиламин. Реагент 3: сульфированный 4-деметил-13- диоксоланилдауномицинон. Условия реакции: диметилформамид или диоксан, инертная атмосфера, 40-60°С, 5-18 ч,...
Способ получения производных 2,5 – дихлорфенола

Номер патента: 3485
Опубликовано: 10.06.1996
Авторы: Йозеф Драбек, Манфред Бегер
МПК: C07C 43/225, C07C 41/05
Метки: получения, производных, дихлорфенола, способ
Формула / Реферат:
Использование: в качестве исходных материалов для синтеза обладающих пестицидной активностью бензоилмочевин. Продукт: соединение формулы I CF3CHF-CF2-O-C6H2(2.5-Cl, 4-R) где R-NH2 (I a) или HO-C6H2. Реагент 1: фенол (II) 4-NH2, в котором NH2 = группу предварительно ацилируют. Реагент 2: гексафторпропен: Полученное соединение в случае необходимости (где R означает NH2) обрабатывают фосгеном для получения соединения формулы I, где R означает...
Способ получения производных 1,4 – дигидропиридина

Номер патента: 1563
Опубликовано: 15.12.1994
Автор: Петер Нойманн
МПК: C07D 211/90, A61K 31/44
Метки: производных, дигидропиридина, получения, способ
Формула / Реферат:
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1,4-ДИГИДРОПИРИДИНА общей формулы где R1 - С1-С4-алкил, R2 -С1- С4-алкил или С1-С4-алкокси, R3 - С1-С4-алкил или С1-С4-алкокси, R4 - С1-С4- алкил; R5 - атом водорода или С1-С4-алкокси; Х - кислород или сера, отличающийся тем, что соединения общей формулы где R5 и Х имеют указанные значения, подвергают взаимодействию с соединениями общей формулы R4COCH2COR3, общей формулы R2COCH2COR1, где R1, R2, R3, R4, указанные...
Способ получения метиловых эфиров 2-гетарил-3-метоксиакриловой кислоты

Номер патента: 2103
Опубликовано: 15.06.1995
Авторы: Вивьен Маргарет Энтони, Джон Мартин Клау, Брайен Кеннет Снелл, Пол де Фрейн, Кевин Бьютмент
МПК: C07D 207/08
Метки: метиловых, способ, эфиров, кислоты, получения, 2-гетарил-3-метоксиакриловой
Формула / Реферат:
Изобретение касается гетероциклических соединений, в частности получения метиловых эфиров 2-гетарил-З-метоксиакриловой кислоты формулы I CH3O-CH=C[A]-C[C]OCH3 где А - группа формулы где R1=Н, CN, фенилэтил, [Е]-фенилэтилен, фенилтио, пиридилтио, формил, С1-4 -алкилкарбонил [он может быть замещен Cl], C1-4 -алкоксикарбонил [он может быть замещен Cl], аллилоксикарбонил, фенилацетил, метоксикарбонил-пропионил, этоксикарбонилацетил, фенилпропионил,...
Производные бензимидазола и промежуточные соединения для их получения

Номер патента: 3907
Опубликовано: 16.09.1996
Авторы: Томоюки Кусаба, Масаюки Еномото, Масае Сугано, Дзуниа Такахаси, Масахиро Тамаки, Реи Матсунага
МПК: A01N 43/12, C07D 405/02
Метки: соединения, получения, бензимидазола, промежуточные, производные
Формула / Реферат:
Использование: в сельском хозяйстве в качесве фунгицида. Сущность изобретения: производные бензимидазола ф-л (1 и 2). Соединения ф-л (3 - 5). где R-циано- или тиокарбомоил Q-низший алкил или группа NQ1, Q2, где Q1 и Q2 независимо низший алкил, низший алкил, замещенный фенилом или низший алкил или Q1 и Q2 вместе взятые с атомом азота образуют пятишестичленное азотсодержащее кольцо, которое может включать атом кислорода: Х-водород или галоген;...
Предыдущий патент: Способ получения стерильного, непирогенного раствора антрациклинового гликозида для инъекций
Следующий патент: Устройство для учета результатов опроса общественного мнения
Случайный патент: Способ укрепления проксимального анастомоза при протезировании брюшной аорты