Катализатор для неселективного окисления органических соединений





Формула / Реферат
Катализатор для неселективного окисления органических соединений, содержащий каталитически активный компонент -оксид переходного металла, стабилизирующий компонент - оксид лантана, алюминат переходного металла и носитель - окись алюминия. При этом в качестве переходного металла катализатор содержит оксид меди, или оксид кобальта, или оксиды меди и марганца, оксид лантана нанесен на поверхность носителя так равномерно, что после обработки при 1050°С в течение 6 ч катализатор не проявляет дифракционного максимума на дифракционной картине рентгенограммы, имеющего полуширину, меньшую 1,0 дугового градуса при измерении по двойному углу Дифракции, при следующем содержании компонентов, мас.%: оксид переходного металла 9,52-18,30; оксид лантана 0,43-2,83; алюминат переходного металла 0,07-0,32; носитель остальное. 1 табл.
Текст
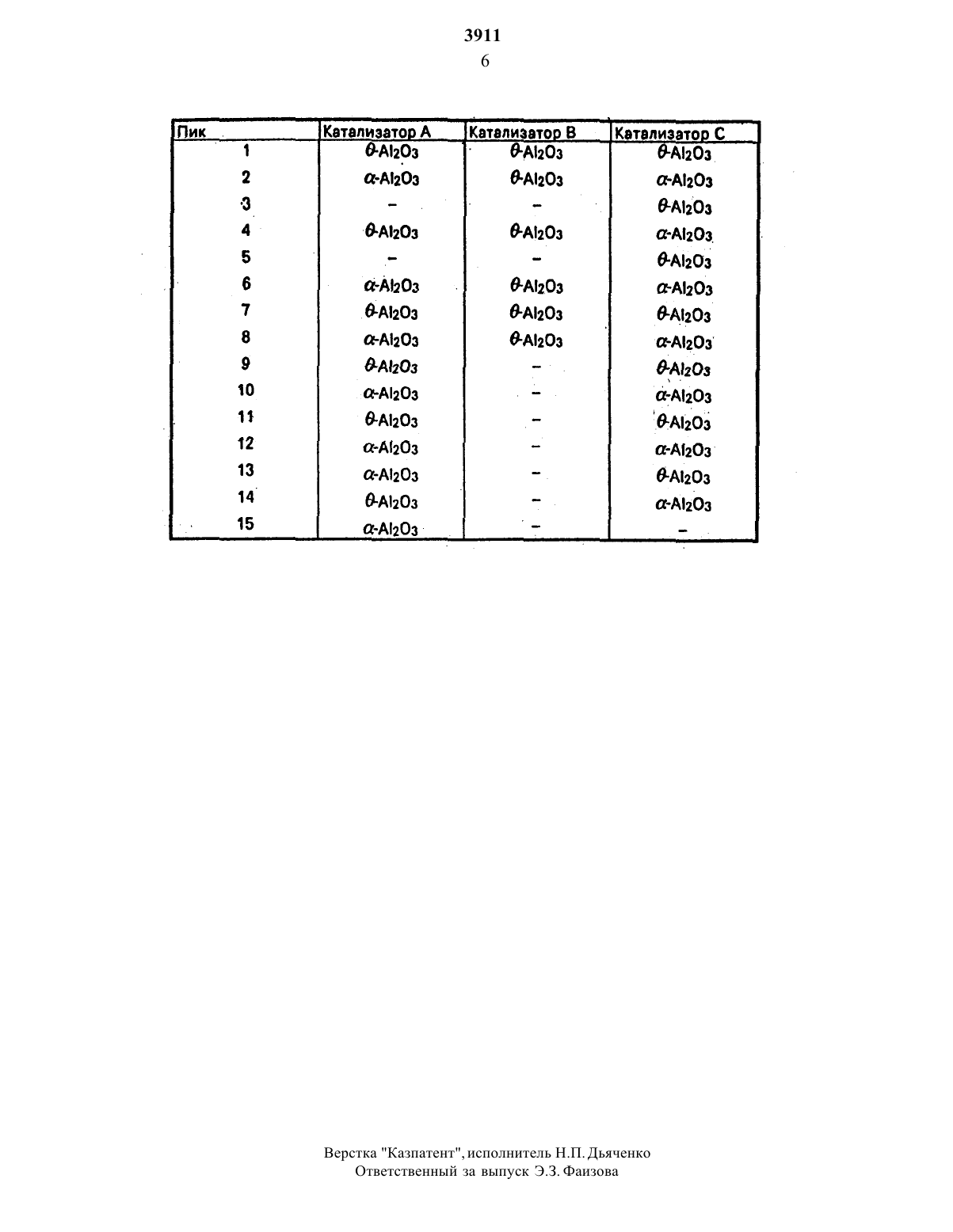
а-АпОз и не показал или по существу не показал линий алюмината меди на дифракционной картине рентгенограммы после нагрева в течение 24 ч при 1000 С. кроме того отсутствовали и линии алюминатов металлов после нагрева в течение 24 чпри 60 ОС в окисляющем газе. Анализ катализатора с использованием электронной микроскопии(селективной поверхностной электронной дифракции) показал кроме того. что в течение этой тепловой обработки ни частицы а-А 2 О 3. ни частицы алюмината меди. имеющие равмеры. превышающие 0.1 мкм. не были обнаружены. То же было обнаружено и для других окислов металлов первых рядов переходных элементов. такихкак кобальт или никель.По-видимому, нанесение стабилизирующего металла или стабилизирующего сое 2динения металла из побочной подгруппы лгруппы З или 4 Периодической таблицы на поверхность носителя приводит к усилению кристаллографического превращения поверхности. так что она не реагирует или по существу больше не реагирует с каталитиче СКИ ЭКТИВНЫМ материалом ИЛИ ЕГО предше- оВ этой связи термин равномерно нанесенный означает, что на единице площади носителя. например на 100 А 2 присутствует по существу равное количество ионов металлов З-й и 4-й групп. Поэтому несущест БЭННО. ЧТОбЫ ПрИСУТСТВОВЗЛФЛОНОСПОЙстабилизирующих ионов металлов. Много меньшее количество ионов необходимо для равномерного распределения по поверхности носителя, и достаточное количество их должно присутствовать. чтобы избежать кристаллографического превращения всей площади поверхности в нежелательнуНевозможно идентифицировать исход НЫЙ НОСИТВЛЬ И стабилизирующее СОВДИНВ(побочных подгрупп) в качестве самостоя тельных соединений. Это скорее всего являя. ется следствием включения ионов металловВ решетку ОКИСИ металла НОСИТЕЛЯ. КОЛИЧС СТВО СОЕДИНЕНИЯ ДОПОЛНИТЕЛЬНОГО ЗПЕМВН та может ИЗМЗНЯТЬСЯ В ШИрОКИХ пределахпри условии равномерного распределения.щий изобретению. может быть получен с помощьюообычных методов нанесения настабилизированный носителыокисла переХОДНОГО МВТЗЛЛЭ. отличающегося ОТ металла стабилизирующего соединения или стабилиэирующего элемента. Подходящими споЯБЛЯЮТСЯ осаждение СИСПОЛЬЗОВЭНИЗМ изменения рН. например путем добавления каустической соли, или с помощью электрохимического процесса с последующим разделением. сушкой и, если это требуется. прокаливанием катализатора.стабилизированный носитель получается путем осаждения на подходящем носителе или адсорбции этим материалом требуемого соединения металла или его предшественника. Согласно одному из ва- ВИЗНТОВ ТЗКОГО СПОСОБЭ ЭТО соединение ме талла наносится путем адсорбции комплекса рассматриваемого иона металла в водном растворе при постоянной величине рН. Такая адсорбция может осуществляться при величинах рН. равных 4-10. при степени адсорбции, которая также определяется выбранной величиной рН.В качестве используемого комплексообразующего агента можно сослаться на обычные комплексообразующие агенты. известные в технике. такие как ЭДТКЭГГК. цитраты. оксалаты и тому подобное.После адсорбции. которая обычно может продолжаться от 0.5 мин до 5 ч. жидкость отделяется от твердого вещества. Этот процесс может быть осуществлен с помощью известного способа. такого как фильтрация. Декантация и центрифугирование. Влажный носитель затем обычно высушивается для удаления жидкости и. если это необходимо. подвергается тепловой обработке. чтобы получить требуемую окисную Форму. Обычно такая тепловая обработкаСогласно другому варианту соединение иона металла может быть нанесено на носитет. из окиси металла с помощью гомогенного осаждения. .- - . .дальнейшая обработка носителя после гомогенного осаждения может осуществляться так. как описано применительно к первому варианту выполнения изобретения.Количество соединения иона переходного металла может зависеть, как указано выше. от выбранной величины рН. если адсорбция комплекса начинается. При гомогенном осаждении степень нанесенияможет быть определена количеством соеди-нения. осадившегося из раствора. Другая возможность изменения степени нанесения состоит в том. что стадия нанесения повторяется два или более раз. Таким образом. очень высокие степени нанесения могут быть получены. хотя в этом может не быть необходимости.Катализатор по изобретению используют в способе неселективного окисления органических соединений. такихкак метан. в также отходящих газов. продуктов сгорания и т.д. Катализатор используется в окислен ной форме.Такие способы особенно важны для беспламенного сжигания углеводородов для генерирования электричества. тепла. энергии и так далее. Изобретение иллюстрируется примерами. - - .П р и м е р 1. 20 гу-АиОз (А 4172. 265 м 2/г. объем пор 1.14 мл/г). продаваемого Нагэпаш В./ были суспендированы в 750 мл деионизованной воды при 30 С. Была установлена величина рН. равная 5. с помощью концентрированной НМ 03.195 г ЕОТА (зтилендиаминтетрауксусная кисло та) были растворены в 50 мл деионизован ной- воды при добавлении по каплям концентрированного аммиака с предотвращением снижения рН ниже 4. 2.69 г Ьа(МОз/з-6 Н 2 О (соответствующие конечному нанесению 5 по весу а 203) были рас творены в 5 мл деионизованной воды иосторожно добавлены по каплям к раствору ЭДТК. Величина рН поддерживалась в интервале 4-7 путем добавления по каплям разбавленного аммиака. Получившийсяраствор был влит в водную суспензию т А 2 О 3. Величина рНснова поддерживаласьравной 5 путем добавления по каплям разбавленной НМ 03. В течение 1 ч суспензияподдерживалась постоянной путем добавления разбавленной НЬЮ ниже поверхнобыла отфильтрована и фильтрат дважды пром ыт по 25 мл деионизованной воды. Несущий материал был затем высушен при 60 С в течение 16 ч. Высушенный несущийматериал был прокален втечение 5.5 ч навоздухе при 550 С. чтобы превратить лантановый комплекс в окисную форму. Несущий материал окончательно содержит 3 по ве су АЬОз. 15 г этого стабилизированного но- сителя были суспендированы в 750 мл деионизованной воды при 30 С 5.16 г Си(МОз)23 Н 20 были растворены в 50 мл деионизованной воды и добавлены к суспензии. Суспензия энергично перемешива 3лась при продувании н под поверхностьжидкости. Величина рН поддерживалась равной 4 с помощью концентрированной НМОз. Путем введения 0.5 н. раствора МаОН(03 мл/мин) под поверхность жидкости величина рН была увеличена до 12. По прошествии 16 ч катализатор был отфильтрован идважды промыт 25 мл деионизованной во ды. Катализатор был высушен в течение 23 ч при 60 С. После нагрева в окисляющем газе в течение 24 ч при 600 С линии алюмината меди не могли быть выявлены в дифракционной картине рентгенограммы. Состав катализатора 0.22 вес. алюмината меди 2.83 .вес. 13203 9.52 вес. СиО остальное А 2 Оз сП р и м е р 2. Каталитическая активность описанного катализатора (1036 по весу Си 0/А 2 Оз) при окислении металла была исследована в реакторе сплотным слоем. Катализатор сжимался придавлении 150 МПа и затем просеивался до тех пор. пока не была получена окончательная фракция 500850 мкм. Реакторанагружался 0.6 г такой просеянной фракции. Газообразная смесь(11, по объему СН 4. 4 по объему 02. 95 по объему М 2) пропускалась над катализатором. Объемная скорость потока составляла 3000 ч. Превращение метана в С 02 и Н 20 наблюдалось уже при 300 С. При 550 С превращение составляло 100. Для того. чтобы исследовать стойкость катализатора. ката лизатор был предварительно обработан впредварительной обработки катализатор был охлажден до комнатной температуры. и реакционная смесь 0 по объему СНА. 4 по объему 02. 95 по объему М 21 была снова пропущена над катализатором. Активность катализатора была еще раз определена. Деактивация не была установлена. Неожидан-но было обнаружено, что активность катализатора быладаже заметно увеличенав результате предварительной обработки. При температуре 460 С превращение уже было равным 100.П р и м е р З. 15 г стабилизированного носителя, способ получениякоторого олисан в примере 1. были суспендированы в 750 мл деионизованной воды при 30 С 6.04 г Со(Юз 026 Н 2 О были растворены в 50 мл деионизованной воды идобавлены к суспензии. Суспензия была энергично перемешана при вдувании азота под поверхность жидкости. Величина рН поддерживалась равной 4.8 с помощью концентрированной НМОз. Путем введения 0.25 М раствора МаОН 0.3 мл/мин) под поверхность жидко сти величина рН была увеличена до 12.5. Попрошествии 16 ч катализатор был отфильтрован и дважды промыт 25 мл деионизованной воды. Катализатор был высушен а течение 23 ч при 60 С. В результате был получен катализатор. содержащий 0.24 вес. алюмината кобальта. 2.83 мас.п Катализвторбыл подвергнуттем же исследованиям. которые описаны в примере 2.дезактивация не была установлена. Посленагрева в окисляющем газе алюминат кобальта не был выявлен на дифракционной картине рентгенограммы.П р и ме р 4. 15 гстабилизированного носителя, способ получения которого ана логичен описанному в примере 1. были сус пендированы в 750 мл деионизованной воды при З 0 С. 75.70 г Си(МОз)2-ЗН 2 О и 5.41 г Мл(МО 3)24 Н 2 О были растворены в 50 мл деионизованной воды и добавлены к сус А пензии. Суспензия была энергично переме шана при вдувании азота под поверхность жидкости. Величина рН поддерживалась(О.3 мл/мин) под поверхность жидкостивеличина рН была увеличена до 12.- По прошествии 16 ч катализатор был отфильтрован и дважды промыт 25 мл деиониэованной воды. Катализатор был высушен в течение 23 ч при 60 АС. В результате был получен катализатор, содержащий 0.09 вес. алюмината меди. 2, 59 вес. 1.3203. 10.10 вес. СиО. 8.20 вес. МпО 2. остальное А 2 О 3 Катализатор был подвергнут тем же ис пытаниям, что и описан в примере 2. деак тивация не была обнаружена. Также после проведения испытаний, которые описаны в примере 1. не были обнаружены алюминаты ни меди. ни марганца.П р и м е р 5. 10 г стабилизированного носителя. способ получения которого аналогичен описанному в примере 1. при нанесении 0,5 по весу .а 2 Оз. были суспендированы в 1.5 л деионизованной воды при 25 С. В качестве электролита были добавлены 7.84 г К 2 ЗО 4. Азот продувалсяплатиновый катод были помещены в суспензию. Величина рН поддерживалась равноймА/см . Затем катализатор был отфильтрован. дважды промыт 25 мл деионизованной воды и высушен в течение 16 ч при 60 С. Был получен катализатор. который содержал 0.32 вес. алюмината меди. 0.47 вес.Стойкость катализатора была исследована таким же образом. как это делалось в примерах 1 и 2. деактивизации или образования алюмината меди не наблюдалось. Было снова обнаружено. что активность при неселективном окислении метана была увеличена в результате преобразовательной обработки при высокой температуре.Стабилизированный носитель был приготовлен путем пропитки 20 г у-А 20 з раствором нитрата лантана таким образом. что получившийся носитель содержал 0,5 повесу АП 2 Оз. Носитель был высушен в течениеодной ночи при 6 ОС и затем прокален в течение 2 ч при 5 О 0 С. Этот носитель затем был пропитан раствором нитрата меди. чтобы свести к минимуму образование алюмината. так что после сушки и прокаливания был получен катализатор. содержащий 10по весу СиО на АП 2 Оз (катализатор А. срав- дВ соответствии со способом. представленнымв примере 1, был получен носитель,который после сушки и прокаливания содержал 0,5 по весу а 2 Оз. С помощью гомогенного осаждения 10 по весу СиО были нанесены на атот носитель (катализатор В. пример 6). Обакатализатора были затем нагреты в течение б ч при 1000 С. Катализатор изучался с помощью рентгеновской дифракции и электронной дифракции. На дифракционной картине рентгенограммы катализатора А были видны сильные пики а-А 2 Оз по соседству с пиками дАЬОз. На дифракционной картине рентгенограммы катализатора В видны только пики д-А 2 О 3. бНа дифракционной картине при электронной дифракции катализатора А пики аА 20 з и Си-А 204 (алюминат меди) наблюдались рядом с пиками д-АЖ 2 О 3. На дифракционной картине при электронной дифракции катализатора В были видны только пики д-А 20 з.ется ТОЛЬКО ТЕРМОСТОЙКИМ. ТОГДЭ как КЗТЗЛИ ЗЗТОР В ЯВЛЯЕТСЯ ТЗКЖВ СТЗбИЛИЗИПОВЗННЫМпротив реакции активного компонента с но сителем. Исходя из мерз. нестабилизированный катализатор был получен путем пропит КИ И ПООКЭЛИВВНИИ нитратом Меди. КЗК ЭТО гописано для катализатора А Была определена реакционная картина на рентгенограмме этого нестабилиэированногопредставлена в таблице П р и м е р 7. По способу примера 1 получен носитель. который после сушки иПосредством однородного нанесения осаждено 10 вес. СиО на этом носителе.ПослепроцедУрЫ прокаливания катализатор был охлажден до комнатной температуры и затем нагрет со скоростью 5 С/мин до 1000 С. Катализатор выдерживался при этой температуре в течение 6 ч. После этойтермообработки катализатор был охлажден до комнатной температуры и анализировался посредством программированного по температуре термографического анализа до 1000 С. 0.200 г этого катализатора было нагрето со скоростью 5 С/ мин до -1000 С втермическом равновесии. В этом экспери менте было показано. чтопримерно 1 (что составляет 0.2 мг) оксида меди. первоначально осажденного на носителе. преврати лись в алюминат меди.После этого эксперимента новый обра 5зец кальцинированного катализатора былподвергнут той же термообработке. котораяденный описанным образом. показал. что на этот раз было образовано 0.29 мг алюмината меди. Третий эксперимент дал образование 0.23 мг алюмината меди. уП р им е р 8. 15 г стабилизированного носителя. приготовленногопо способу. описанному в примере 1. с нагрузкой 15 вес. 1 а 2 О 3. обращены в суспензию в 750 мл деионизированной воды при З 0 С 5.16 г Си(МО 3)2- ЗН 2 О было растворено в 50 мл деионизированной воды и добавлено к суспензии. Суспензию энергично размешивали при продуве над поверхностью азота На. Величина рН 4 была установлена с помощью концентрированной азотной кислоты НМОз. вводом 0.5 М раствора МаОН(0.3 мл/ мин) под поверхность жидкости была установлена величина рН 12. После 16 ч катализатор отфильтровали и дважды промыли в 25 мл деионизированной воды. Катализатор высушивали при 60 С в течение 24 ч. После нагревания в окисляющем газе в течение 24 ч при Б 00 С катализатор прошел предварительную обработку в потоке азота при 10 ООС в течение б чцпосле этой предварительной обработки состав катализатора был такой 17 вес. алюмината меди. 13.45 вес. .а 2 Оз. 9.83 вес. оксида меди и 76.55 вес. АЬОэ.При м е р 9. В этом примере два образца уокиси алюминия (марки Энгель-хард А 4172) было пропитано 4.3 вес. лан тана. Первый образец. обозначенный Катализатор 1. был пропитан по известному из уровня техники методом по влагоемкости. Предшественником стабилизирующего оксида был нитрат лантана. Второй образец. обозначенный катализатор- 2 был приготовлен поЭти образцы катализатора исследовали на сканирующем электронном микроскопеи проводили анализ по элементам. На электронной микрофотографии катализатора 1 белые пятна показывают присутствие оксида лантана. Добавочно производилось сканирование элементного анализа лантана вдоль белой линиина микрографии. Там, где белая линия касается белогопятна. обнаруживается лантан в анализе на элемент. Для катализатора 2 электронная микрофотография не показывает белых пятен на носителе. что подтверждает очень равномерное распределение оксида лантана по поверхности носителюВ этом случае нельзя было провести анализ на элементы сканированием. так как разрешающая способность оборудования для анализа недостаточна для обнаружения частиц оксида лантана. Частицы оксида лантана были длиной примерно 10 мкм. а шириной 1.5 мкм. . добавочная работа по характеристикам проводилась с помощью электронного микраскопа. работающего на пропускание. С помощью этой технологии был определен главный размер частиц. он оказался приОмерно 5 А. Анализ микрофотографий показал Примерно 311 частиц оксида лантана наКатализатор для неселективного окисления органических соединений. содержащий каталитически активный компонент оксид переходного металла. стабилизирую ЩИЙ КОМПОНЕНТ ОКСИД ЛЭНТЗНЗ. алюминатцелью ПОВЫШЕНИЯ термической И ХИМИЧет ской устойчивости катализатора. в качестве оксида переходного металла катализатор содержит оксид меди. или оксид кобальта,илиоксиды меди и марганцаюксид лантана нанесен на поверхность носителя так рав. номерно. что после обработки при температуре .1050 С в течение 6 ч катализатор непроявляет дифракционного максимума на дифракционной картине рентгенограммы. имеющего полуширину. меньшую. чем 1.0у ДУБОВОЙ градус при ИЗМВРЗНИИ ПО ДВОЙНОМУ
МПК / Метки
МПК: B01J 23/10, B01D 53/36
Метки: катализатор, неселективного, органических, соединений, окисления
Код ссылки
<a href="https://kz.patents.su/6-3911-katalizator-dlya-neselektivnogo-okisleniya-organicheskih-soedinenijj.html" rel="bookmark" title="База патентов Казахстана">Катализатор для неселективного окисления органических соединений</a>
Катализатор для избирательного окисления серусодержащих соединений

Номер патента: 2752
Опубликовано: 15.12.1995
Авторы: Питер Хильдегардус Бербен, Джон Вильхельмус Гэс
МПК: B01J 23/86, C01B 17/04
Метки: окисления, катализатор, избирательного, серусодержащих, соединений
Формула / Реферат:
Катализатор для избирательного окисления серусодержащих соединений, в частности сероводорода, с образованием элементарной серы, включающий носитель, поверхность которого, доступная для контакта с газовой фазой, не обладает щелочными свойствами в условиях реакции, и каталитически активный компонент - окись железа или окись железа и хрома. Нанесенный на поверхность носителя, причем величина удельной поверхности катализатора составляет менее 20 м2...
Катализатор для глубокого окисления углеводородов

Номер патента: 3600
Опубликовано: 10.06.1996
Авторы: Гладун Галина Георгиевна, Касымбекова Дария Азыкановна, Космамбетова Гульнара Радиевна, Соколова Людмила Антоновна
МПК: B01D 53/36, B01J 23/26, B01J 21/04...
Метки: углеводородов, окисления, глубокого, катализатор
Формула / Реферат:
Изобретение относится к каталитической химии, в частности, к катализаторам для глубокого окисления углеводородов и может быть использовано в химической и нефтехимической промышленности. Катализатор для глубокого окисления углеводородов включает активную фазу в виде соединений магния и хрома и оксида алюминия. Катализатор получен в режиме самораспространяющегося высокотемпературного синтеза из экзотермической смеси при следующем соотношении...
Способ очистки газов от сернистых соединений

Номер патента: 3085
Опубликовано: 15.03.1996
Автор: Робер Вуарэн
МПК: C01B 17/04, B01D 53/34
Метки: сернистых, способ, очистки, газов, соединений
Формула / Реферат:
Изобретение относится к процессам каталитической очистки газов от сернистых соединений и позволяет повысить стабильность процесса при сохранении активности катализатора на высоком уровне. Для осуществления очистки ведут контактирование газа, содержащего сернистые соединения, с алюмокобальтмолибденовым катализатором для гидрирования сернистых соединений до сероводорода, а затем газ пропускают через катализатор, содержащий окислы железа или...
Катализатор для получения высокомолекулярных цис- этиленовых изомеров и способ его приготовления
Номер предварительного патента: 1358
Опубликовано: 15.12.1994
Авторы: Пак Алла Михайловна, Картоножкина Ольга Ивановна, Издебская Гульнара Тастемировна
МПК: B01J 23/04, C07C 33/02
Метки: способ, этиленовых, цис, приготовления, катализатор, изомеров, высокомолекулярных, получения
Формула / Реферат:
Изобретение относится к способам приготовления катализатора для органического синтеза и может быть использовано при получении цисолефиновых соединений, являющихся промежуточными продуктами синтеза феромонов насекомых-вредителей, в частности при каталитическом синтезе цисэтиленовых углеводородов, альдегидов, спиртов. Предполагаемое изобретение позволяет повысить активность катализатора гидрирования за счет определенного способа приготовления и...
Катализатор для дегидрирования углеводородов

Номер патента: 2750
Опубликовано: 15.12.1995
Авторы: Фредерик Ферслуйс, Гейнц Циммерманн
МПК: C07C 5/333, B01J 21/06, B01J 23/26...
Метки: катализатор, дегидрирования, углеводородов
Формула / Реферат:
Сущность изобретения: катализатор содержит оксид хрома 25% БФ Сr2О3; соединение щелочного и/или щелочноземельного металла, в пересчете наоксид, 0.7-4,7%; диоксид циркония 0,9%, БФ ZnO2 и носитель-оксид алюминия - остальное, БФ Аl2О3. Оксид алюминия пропитывают раствором, содержащим оксид хрома и оксиацетат циркония. Сушат сначала в вакууме, затем в атмосфере воздуха.Кальцинируют в атмосфере воздуха. Затем пропитывают раствором соединения...
Предыдущий патент: Шахтная сушилка для сыпучих материалов
Следующий патент: Плазменная горелка
Случайный патент: Многодвигательный электропривод переменного тока